Cardiac amyloidosis in a Swiss autopsy cohort – distribution and clinical relevance
DOI: https://doi.org/https://doi.org/10.57187/s.4541
Albert Baschonga*,
Sara Ersözlübcd*,
Frank Ruschitzkabeg,
Andreas J. Flammerb,
Christoph A. Meierfh,
Zsuzsanna Vargaa,
Holger Mochag,
Umberto Maccioa
a Department of Pathology and Molecular
Pathology, University Hospital Zurich, Zurich, Switzerland
b Department of Cardiology, University
Hospital Zurich, Zurich, Switzerland
c Department of Nuclear Medicine, Cardiac
Imaging, University Hospital Zurich, University of Zurich, Zurich, Switzerland
d Cardiovascular Imaging Research Center,
Massachusetts General Hospital, Harvard Medical School, Boston, MA, United
States
e Centre for Translational and Experimental
Cardiology (CTEC), Department of Cardiology, University Hospital Zurich,
University of Zurich, Zurich, Switzerland
f Department of Internal Medicine,
University Hospital Zurich, Zurich, Switzerland
g Medical Faculty, University of Zurich,
Zurich, Switzerland
h Faculty of Medicine, University of Geneva,
Geneva, Switzerland
* These authors contributed equally to this manuscript
Summary
AIMS: Cardiac amyloidosis (CA) characterised by myocardial amyloid accumulation is
likely underdiagnosed. The
distribution and extent of myocardial amyloid deposits remain unclear. With the
emergence of disease-modifying drugs for ATTR and AL amyloidoses, early
detection has become increasingly important. We aim to determine the frequency,
clinical relevance and distribution of amyloid subtypes in cardiac amyloidosis
in an autopsy cohort.
METHODS: We retrospectively analysed
consecutive unselected adult autopsies with cardiac amyloidosis over 10 years (January
2014 – December 2023). Two pathologists applied a biventricular
semi-quantitative scoring system for interstitial and vascular amyloid
deposits. Histopathological findings were correlated with ante mortem clinical
data.
RESULTS: Cardiac amyloidosis was found in
104 of 1972 autopsies (5%) with 91% neither diagnosed nor suspected ante mortem
based on documentation in digital medical records. Ninety-eight patients (94%)
had amyloid transthyretin-cardiac amyloidosis (ATTR-CA) and six (6%) amyloid
light chain-cardiac amyloidosis (AL-CA). AL-CA patients were younger than
ATTR-CA patients (mean ± SD: 73.2 ± 15.3 vs 84.2 ± 8.1, p
= 0.006) and systemic amyloidosis was more frequent (100% vs 38%, p = 0.003).
Female patients (40.4%) were significantly older (mean ± SD: 85.8 ± 8.1
years) than males (82.0 ± 9.2 years, p = 0.23), and male sex was
associated with clinical suspicion and diagnosis (88.9% in males vs 11.1% in
females, p = 0.06). A high vascular amyloid score correlated with systemic
amyloidosis (left ventricle, p = 0.003; right ventricle, p = 0.013). Right
ventricular amyloid burden was strongly linked to clinical suspicion and
detection (p = 0.001).
CONCLUSIONS: Our autopsy analysis found
that most cardiac amyloidosis cases were undiagnosed ante mortem, especially
ATTR-CA in older patients with less systemic involvement. Underdiagnosis was
more pronounced in females. Our findings suggest that high vascular amyloid
burden contributes to systemic amyloidosis and links right ventricular amyloid
to clinical suspicion and detection.
Abbreviations
- AA-CA
-
serum amyloid A-cardiac amyloidosis
- AL-CA
-
amyloid
light chain-cardiac amyloidosis
- ATTR-CA
-
amyloid transthyretin-cardiac amyloidosis
- ATTRv
-
variant form of amyloid transthyretin
- ATTRwt
-
wild-type amyloid transthyretin
- CA
-
cardiac amyloidosis
- ESC
-
European Society of Cardiology
Introduction
Cardiac amyloidosis (CA) is a storage
disease characterised by extracellular deposition of insoluble misfolded amyloidogenic
proteins [1, 2]. Clinically, it is frequently identified at a late stage,
particularly ATTR amyloidosis. As a progressive disorder, it carries a poor
outcome if left untreated [3]. Currently, more than 40 proteins are known to be
capable of aggregating as amyloid in vivo, of which nine have been detected in
the heart so far [4]. The most frequent forms are amyloid transthyretin-cardiac
amyloidosis (ATTR-CA) and amyloid light chain-cardiac amyloidosis (AL-CA) [5–7].
The early identification of ATTR and AL amyloidoses is of paramount importance in
view of the availability of disease-modifying drugs such as tafamidis for
ATTR-CA and the anti-CD38 monoclonal antibody
daratumumab for AL-CA [5, 8].
ATTR-CA, predominantly acquired wild-type amyloid
transthyretin (ATTRwt), is closely linked to ageing, invariably affects the
heart and has a median survival of 57 months from diagnosis [9]. Conversely, hereditary
forms of transthyretin amyloidosis (ATTRv) represent a less common and
heterogeneous group, which frequently exhibits extracardiac
manifestations, and a variable penetrance and prognosis based on the specific
mutation involved [10–13]. Studies indicate that ATTR amyloidosis is a
frequently overlooked cause of increased left ventricular wall thickness (LVWT),
particularly in individuals aged 65 or older, including those with hypertrophic
cardiomyopathy (5%), heart failure with preserved ejection fraction (HFpEF) (13%)
or severe aortic stenosis undergoing transcatheter aortic valve implantation (TAVR)
(16%) [14–17].
AL amyloidosis is caused by a B cell clone
producing an amyloidogenic light chain and can affect all organs except for the
central nervous system. The heart is affected in up to 70% of AL amyloidosis cases
[1, 18]. The overall median survival is 24
months from diagnosis, dropping to 6 months if untreated heart failure is
present at diagnosis [19, 20]. Acquired AA amyloidosis is a less common form of
amyloidosis
caused by the overproduction and accumulation of the acute-phase protein serum
amyloid A that can be highly expressed in patients with chronic inflammation,
cancers or (auto)inflammatory diseases [21]. Cardiac involvement was found
in 5% of cases diagnosed with AA amyloidosis and was associated with a median
survival of 133 months [1, 21].
In a recent study, our research group
looked at the frequency of undiagnosed diseases in autopsies and we found that
up to 8% of all autopsies conducted at our institution had cardiac amyloidosis
that was clinically undiagnosed prior to death in patients older than 18 years
at death [22]. The true epidemiology of cardiac amyloidosis remains uncertain
as not all deceased patients undergo postmortem examination
[23, 24]. Various studies indicate that cardiac amyloidosis is more prevalent
than previously assumed, particularly among elderly patients [23, 25–27]. In
fact, postmortem investigations have reported cardiac amyloidosis in 22–25% of
patients aged over 80 years [25] and in 14–32% of those aged over 75 years [26].
A single-centre study from Italy has recently reported a 43% incidence of cardiac
amyloidosis (50% ATTR and 50% AL) in hearts from patients aged 75 years or over
[27]. The presence of cardiac amyloidosis significantly correlated with
age, hypertension, chronic kidney disease, coronary artery disease and
hypertensive cardiomyopathy in our previous study [22].
While previous studies in Japanese and
Italian populations have explored select clinicopathological correlations, none
has specifically assessed the association between ante mortem clinical detection
and the extent of interstitial or vascular amyloid deposition [27–29]. The aim
of the present study was to characterise the frequency of amyloid subtypes in a
Swiss autopsy cohort and to investigate the histological burden of cardiac
amyloidosis in patients with a missed diagnosis during life, focusing on
potential links with clinical recognition. To our knowledge, this is the first
study to examine an autopsy-confirmed cardiac amyloidosis cohort in
Switzerland, offering new insights into diagnostic gaps and their pathological
correlates.
Materials and methods
Patient cohort
We conducted a retrospective review of 1972
reports of unselected consecutive whole-body autopsies of adults performed at
the Department of Pathology and Molecular Pathology of the University Hospital
of Zurich, Switzerland, between 1 January 2014 and 31 December 2023. Autopsy
reports were screened to identify patients with cardiac amyloidosis. Each autopsy
had been performed by a pathology resident under the supervision of a
board-certified pathologist according to a standardised protocol as previously
described [22, 30]. This protocol also includes the heart autopsy and the routine
histological analysis of four samples of myocardial tissue from various
anatomical regions (anterior and posterior cardiac wall, septum and right
ventricle). Presence of amyloidosis was identified on routine Haematoxylin
& Eosin and Elastin-van Gieson staining and verified by Congo Red staining. On Haematoxylin
& Eosin staining, it appears as an amorphous eosinophilic substance within
the interstitium and stained with Congo Red shows a yellow-green birefringence
under polarised light [31]. All cases with evidence of cardiac amyloidosis underwent
additional staining with a standard immunohistochemical panel (ATTR, AL and AA) to
classify the
type of amyloidosis, following the
recommendations of Linke [32].
Consent for the autopsy (either by
the relatives or in a few cases by the deceased patient’s will) was obtained for
all cases.
The study was conducted in compliance with Swiss federal research regulations
and received approval from the institutional review board and Cantonal Ethics Committee
Zurich (Identifier BASEC-Nr. 2024-01760).
Immunohistochemistry
Amyloid immunohistochemistry was performed
using amYmed (ATTR, AL) and Dako (AA) antibodies (see appendix table S1). Signal
amplification and visualisation were done with the OptiView DAB IHC Detection
Kit (Ventana Medical Systems) for ATTR and AL and with the IHC Refine Kit
(Biosystems) for AA, following the manufacturers’ protocol. Slides were
counterstained with haematoxylin, sequentially dehydrated and coverslipped for
microscopic evaluation. AL was only considered detected if both antibodies (HAR
and ULI/LAT) appeared positive.
Amyloid scores and clinical data
Clinical data were obtained from referral
documents for autopsies and electronic medical records. Patients were
classified as having been diagnosed or suspected of cardiac amyloidosis ante mortem
based on the available clinical information. For patients who died at our
institution, full electronic medical records – including cardiology notes and
imaging reports – were reviewed. In patients for whom no electronic medical
records were available (i.e. externally referred cases), classification was
based solely on the referral documents submitted for autopsy. The
classification terminology reflects the wording used in the original clinical
documentation: patients in whom amyloidosis was discussed as a differential
diagnosis or was under investigation and explicitly phrased as “suspected” were
categorised as suspected, while those in whom the diagnosis was documented as
established were categorised as diagnosed. This approach was chosen to reflect
how patients were clinically classified ante mortem.
Patients with available cardiological
documentation within one year prior to death were included in a subgroup
analysis of clinical manifestations.
Descriptive statistics were used to analyse
the frequency of cardiac amyloidosis. The prevalence of cardiac amyloidosis was
calculated as the proportion of cases with amyloid deposits. The causes of
death were determined from the autopsy reports. The categories were based on
their primary affected system or pathological process in relation to the clinical
context. Cases in which the amyloidosis was directly related to the cause of
death were additionally noted.
We developed a semi-quantitative scoring
system to assess the extent of amyloidosis in the myocardial tissue of the left
and right ventricles, as well as in the myocardial vessels of the left and
right ventricles. On Haematoxylin & Eosin and immunohistochemistry slides,
the percentage of amyloid deposition was quantified in relation to the surface
area of the tissue sample resulting in myocardial scores from 1 to 10 (table
1). The affected vessels were quantified in relation to the total number of
vessels present on the sample, resulting in vessel scores from 1 to 4 (table 1).
Table 1Scoring system to evaluate the extension of amyloidosis in
myocardial tissues and in myocardial vessels.
| Score |
Extent of amyloidosis |
|
Percentage of myocardium containing amyloid |
| 0 |
0% |
| 1 |
1–10% |
| 2 |
11–20% |
| 3 |
21–30% |
| 4 |
31–40% |
| 5 |
41–50% |
| 6 |
51–60% |
| 7 |
61–70% |
| 8 |
71–80% |
| 9 |
81–90% |
| 10 |
91–100% |
|
Percentage of
myocardial vessels containing amyloid |
| 0 |
0% |
| 1 |
1–10% |
| 2 |
10–50% |
| 3 |
50–90% |
| 4 |
90–100% |
| 5–10 |
Not applicable |
Two experienced pathologists (AB and UM) independently
evaluated the histology of myocardial tissue from cases with cardiac
amyloidosis. The myocardial and vascular scores of the left and right
ventricles were assessed for each case, both on Haematoxylin & Eosin and on
immunohistochemistry. In the event of any discrepancies, a joint evaluation was
conducted in order to reach a consensus on the score.
Statistical analyses were performed using
SPSS version 29.0.1.1 (IBM Corp, Armonk, New York, USA). The extent of amyloidosis
was correlated with demographic variables (age and
sex) and with other clinicopathological
variables (presence of systemic vs cardiac-only amyloidosis, amyloidosis type)
using Kendall’s tau-b test. The closer the correlation coefficient (τb) is to 1
or −1, the stronger the correlation between the variables is assumed to
be [33].
Binary
variables were compared with each other using Fisher’s exact test. Given the exploratory
nature of these analyses, no adjustment for
multiple testing was performed. Assessment for statistical significance in the
subgroup analysis for clinical data was evaluated using the independent T-test
for normally distributed values and the Mann-Whitney U test for non-normally
distributed values. Results were considered statistically significant when the p-value
was less than 0.05. Figures were created using GraphPad Prism 10.3.1.
Results
Clinicopathological correlation
Cardiac amyloidosis was identified in 104 of
1972 autopsies (5.3%). Among these cases, the diagnosis was made at the time of
autopsy in 95 of 104 patients (91.3%), while in six patients (5.8%) cardiac
amyloidosis had been diagnosed prior to death. Cardiac amyloidosis was
suspected prior to death but confirmed only at autopsy in 4 patients (2.9%). Females
were slightly older, with a mean ± SD age of 85.8 ± 8.1 years vs 82.0
± 9.2 years in males.
Table 2 details the types and distribution of cardiac and systemic amyloidoses
among the patients. Ninety-eight patients (94.2%) were diagnosed with ATTR amyloidosis,
while only 6 (5.8%) had AL amyloidosis. There was no AA amyloidosis. The
majority of patients in both subgroups were male (59.2% in ATTR-CA and 66.7% in AL-CA).
Among
patients with AL-CA, 66.7% (4/6 patients) received an ante mortem diagnosis,
with no cases classified as suspected, whereas 5.1% (5/98 patients) of ATTR-CA
patients were clinically identified (2 diagnosed, 3 suspected). Relevant
imaging findings, including echocardiography and, in two cases, cardiac
magnetic resonance imaging (CMR), that contributed to the clinical suspicion or
diagnosis are summarised in appendix table S2. One patient had cardiac
amyloidosis confirmed histologically through myocardial biopsy performed during
emergency surgery. None of the patients underwent 99mTc-DPD scintigraphy. In
three patients, classification as diagnosed vs suspected vs undiagnosed was
based solely on the referral documents provided with the autopsy request.
Table 2Types and distribution of amyloidosis in patients with cardiac
amyloidosis (n = 104).
| Variable |
n (%) |
| Amyloidosis distribution |
Only cardiac |
60 (57.7%) |
| Systemic |
44 (42.3%) |
| Amyloidosis type |
ATTR-CA |
98 (94.2%) |
| AL-CA |
6 (5.8%) |
| Cardiac amyloidosis distribution |
Myocardial only |
41 (39.4%) |
| Vascular only |
22 (21.1%) |
| Combined |
41 (39.4%) |
Notably, AL-CA cases demonstrated a
significantly stronger association with clinical suspicion or diagnosis
compared to ATTR-CA (τb = 0.489, p <0.001, n = 104). A statistically
significant correlation was observed between patient age at death and amyloidosis
type: patients with AL-CA (mean ± SD age: 73.2 ± 15.3 years) were
found to be significantly younger than those with ATTR-CA (84.2 ± 8.1
years), as illustrated in figure 1A.
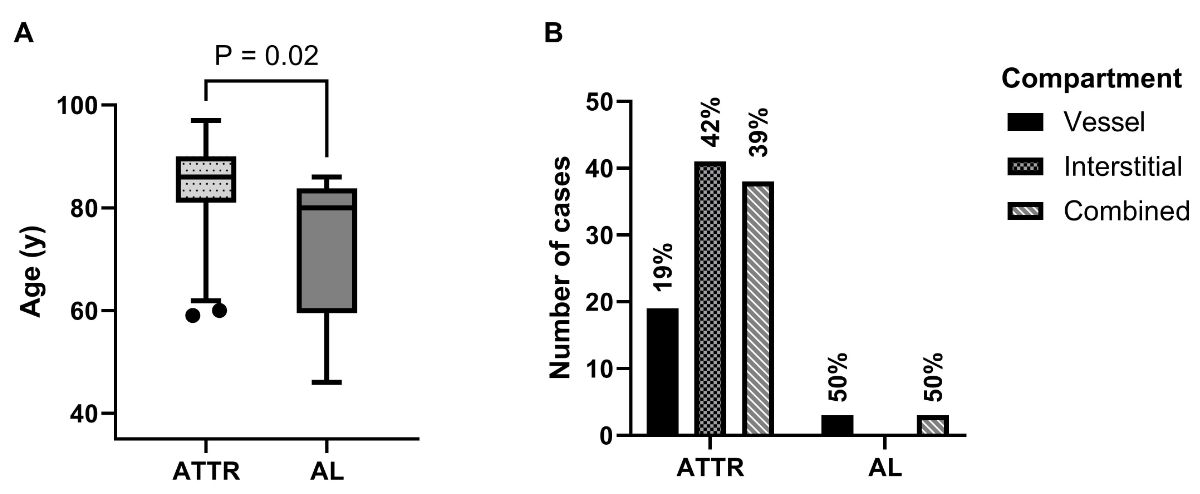
Figure 1(A): Age
distribution of cases classified according to amyloidosis type; whiskers at 2.5 and
97.5 percentiles (p-value calculated with
Mann-Whitney U test). (B): Cases
classified according to the type and distribution of myocardial amyloid
deposits. Percentages of cases by type. AL: amyloid light chain-cardiac
amyloidosis; ATTR: amyloid transthyretin-cardiac amyloidosis.
Sixty of 104 patients (57.7%) exhibited
solely cardiac amyloidosis, while 44 cases (42.3%) presented with systemic
amyloidosis (see table 2), which is defined as amyloidosis involving multiple
organ systems [31]. Patients with AL-CA were found to have a higher prevalence
of systemic amyloidosis than ATTR-CA patients: 100% in AL (n = 6) vs 38.8% in ATTR
(n = 38).
The analysis of clinical information
obtained prior to death revealed that the majority of patients had arterial
hypertension (86.2%) and coronary heart disease (54.6%), while the “red flag”
of hypotension or normotension in previously hypertensive patients was present
in 23.6%. The diagnosis of heart failure with a left ventricular ejection
fraction (LVEF) below 50% was made in 43.6% of patients and 21.7% of patients
were diagnosed with “hypertensive cardiomyopathy” or heart failure with
preserved ejection fraction. Chronic kidney disease was found in 49.1%, atrial
fibrillation was present in 45.5%, AV conduction disease found in 27.3% and
aortic valve stenosis in 29.1%. No statistically significant differences in
cardiac manifestations could be detected between ATTR-CA and AL-CA (see table
3). Among the non-cardiac findings (see appendix table S3), macroglossia (p <0.001),
monoclonal gammopathy of undetermined significance (MGUS) (p <0.001) and haemorrhagic
strokes (p = 0.02) were significantly more frequent in AL-CA than
in ATTR-CA. Interestingly, the red flags lumbar spinal stenosis and
bilateral carpal tunnel syndrome were only rarely found (7.3% and 3.6%,
respectively).
Table 3Patient characteristics and cardiological findings.
| Criteria |
ATTR-CA |
AL-CA |
All |
p-value |
| Total number of patients, n (%) |
98 (94.2%) |
6 (5.8%) |
104 (100%) |
|
| Sex, n (%) |
Male |
58 (59.2%) |
4 (66.7%) |
62 (59.6%) |
|
| Female |
40 (40.8%) |
2 (33.3%) |
42 (40.4%) |
|
| Cardiovascular risk factors, n (%) |
Information available |
83 (84.7%) |
4 (66.7%) |
87 (83.7%) |
|
| Arterial hypertension |
73 (88.0%) |
2 (50%) |
75 (86.2%) |
0.06 |
| Tobacco |
23 (27.7%) |
1 (25%) |
24 (27.6%) |
0.90 |
| Adiposity |
21 (25.3%) |
2 (50%) |
23 (26.4%) |
0.30 |
| Dyslipidaemia |
22 (26.5%) |
0 (0%) |
22 (25.3%) |
0.95 |
| Type 2 diabetes mellitus |
15 (18.1%) |
1 (25%) |
16 (18.4%) |
0.73 |
| Family history for cardiovascular disease |
6 (7.2%) |
0 (0%) |
6 (6.9%) |
0.06 |
| Cardiological findings/diagnosis prior to death as
stated in the reports, n (%) |
Information available |
51 (52.0%) |
4 (66.7%) |
55 (52.9%) |
|
| Hypotensive or normotensive (previously
hypertensive) |
11 (21.6%) |
2 (50%) |
13 (23.6%) |
0.20 |
| Hypertrophic cardiomyopathy |
4 (7.8%) |
0 (0%) |
4 (7.3%) |
0.56 |
| Hypertensive cardiomyopathy / heart failure with preserved ejection fraction |
20 (39.2%) |
2 (50%) |
22 (40.0%) |
0.67 |
| Heart failure with mildly reduced ejection fraction |
10 (23.3%) |
0 (0%) |
10 (21.7%) |
0.33 |
| Heart failure with reduced ejection fraction |
12 (27.9%) |
2 (50%) |
14 (30.4%) |
0.25 |
| Coronary artery disease |
28 (54.9%) |
2 (50%) |
30 (54.6%) |
0.85 |
| Aortic valve stenosis |
16 (31.4%) |
0 (0%) |
16 (29.1%) |
0.19 |
| Aortic valve prosthesis (transcatheter aortic valve
replacement or surgical replacement) |
11 (21.6%) |
0 (0%) |
11 (20%) |
0.30 |
| Atrial fibrillation |
23 (45.1%) |
2 (50%) |
25 (45.5%) |
0.85 |
| Atrioventricular conduction disease |
14 (27.5%) |
1 (25%) |
15 (27.3%) |
0.92 |
| Pacemaker placement |
11 (21.6%) |
2 (50%) |
13 (23.6%) |
0.20 |
| Implantable cardioverter-defibrillator or cardiac
resynchronisation therapy |
2 (3.9%) |
0 (0%) |
2 (3.6%) |
0.69 |
Correlation to
semi-quantitative amyloidosis scores
Myocardial samples of the left ventricle
were available in all 104 patients and of the right ventricle in 99 patients.At autopsy,
41 patients exhibited
interstitial myocardial-only amyloidosis (39.4%), whereas 22 patients had
vascular-only amyloidosis (21.2%) and the remaining 41 patients had combined
vascular and interstitial myocardial amyloidosis (39.4%) (see table 2 and figure
1B). Figure 2 depicts the distribution of cases based on the extent of
interstitial amyloid deposition in the myocardial tissue and the extent of the
vascular amyloid deposition. Figure 3 presents examples of varying degrees of
interstitial and vascular amyloid deposition.
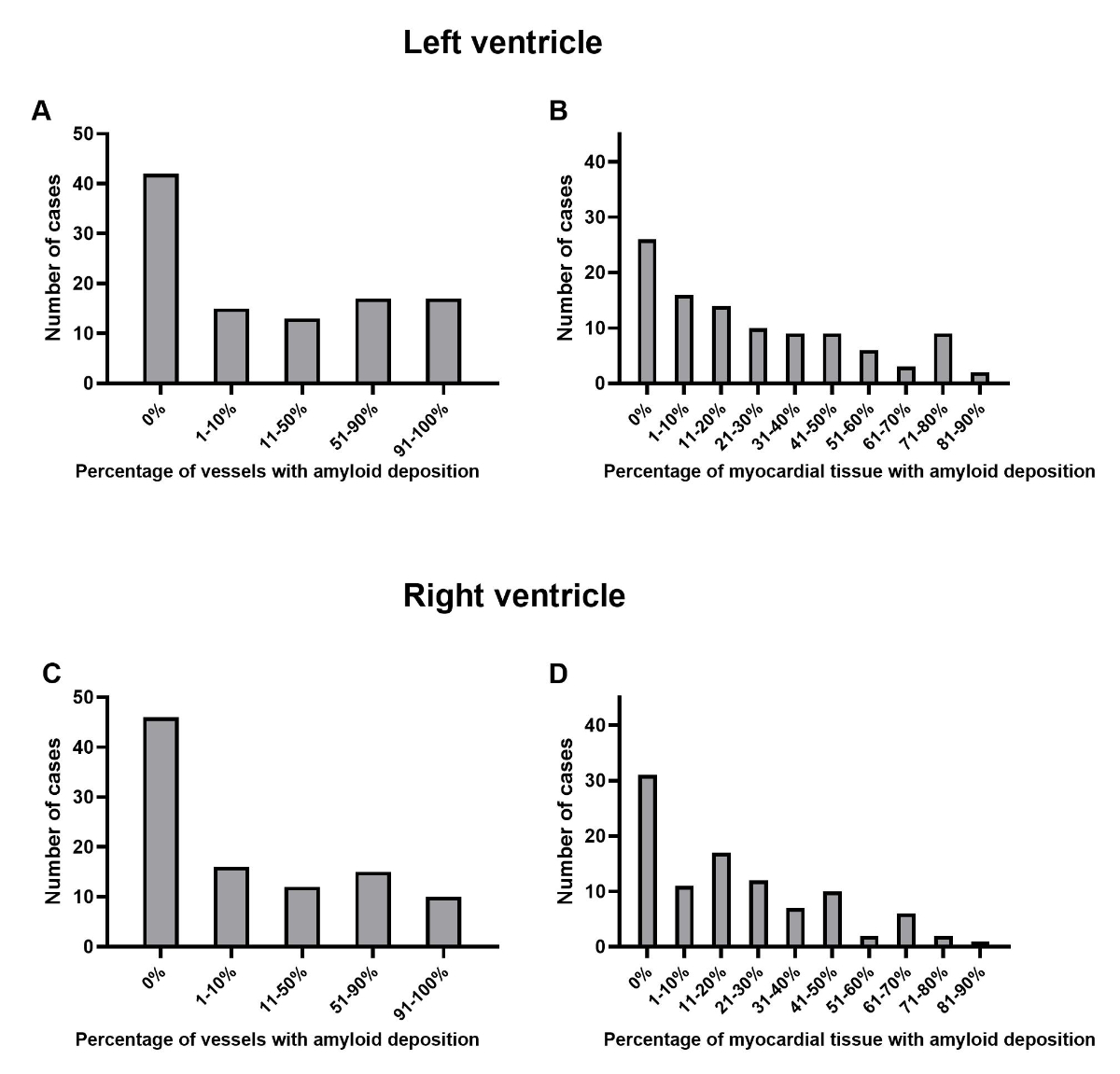
Figure 2Distribution of cases depending on the location and extent of
amyloid depositions. (A and C): Percentage of myocardial vessels with amyloid
deposition categorised by vascular score in the left (A) and right (C) ventricle.
(B and D): Percentage of interstitial amyloid deposition in relation to the
myocardial tissue surface categorised by myocardial interstitial score in the
left (B) and right (D) ventricle.
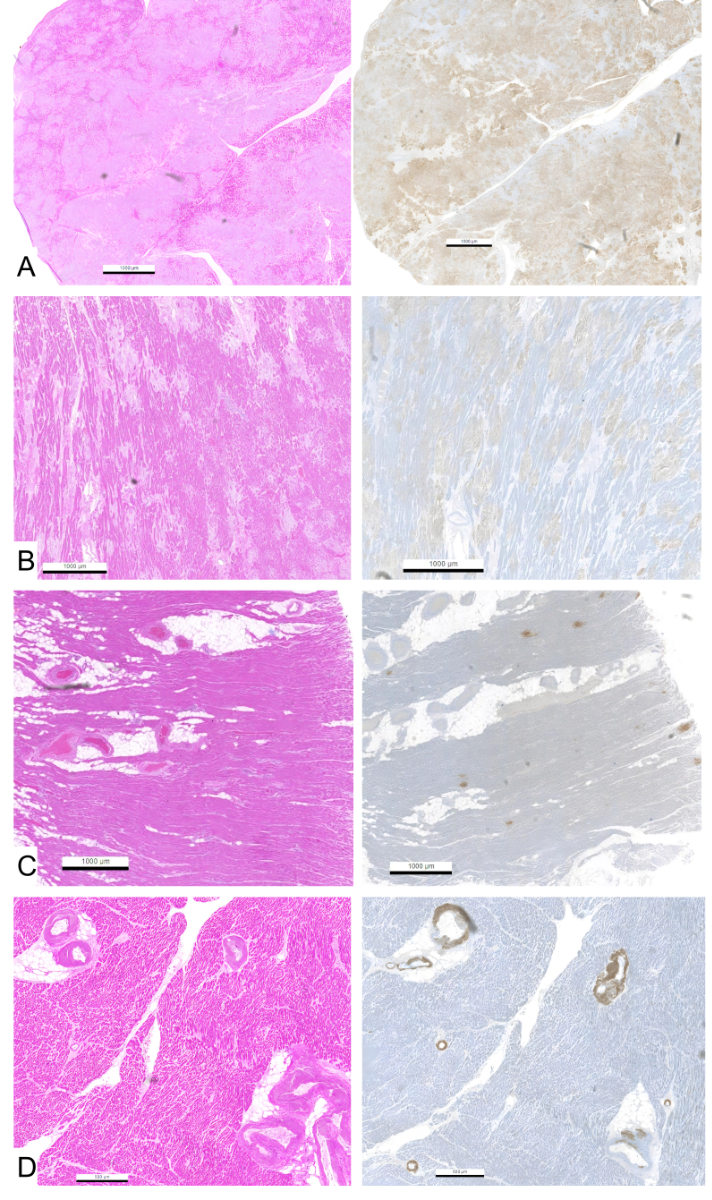
Figure 3Haematoxylin & Eosin slides showing different percentages of
interstitial (A–C) and vascular (D) amyloid deposits with respective score and
corresponding ATTR immunohistochemistry. (A): Extensive interstitial amyloid
deposits in >90% of the myocardial surface, resulting in a myocardial score
of 9. (B): Interstitial amyloid deposits in 50–60% of the myocardial surface,
resulting in a myocardial score of 5. (C): Interstitial amyloid deposits in
under 10% of the myocardial surface, resulting in a myocardial score of 1. (D):
Amyloid deposits shown in >90% of myocardial vessels, resulting in a vessel
score of 4.
A significant correlation was identified
between the vascular score and the presence of systemic amyloidosis in both the
left and right ventricles (left: τb = 0.261, p = 0.003, n = 104; right: τb = 0.229,
p = 0.013, n = 99). However, when focusing specifically on ATTR-CA, this
relationship was evident only in the left ventricle (τb = 0.248, p = 0.007, n =
94), whereas no significant association was observed in the right ventricle (τb
= 0.177, p = 0.06, n = 94).
Additionally, clinically confirmed or
suspected cardiac amyloidosis was significantly associated with an increased
interstitial myocardial score in both the left (τb = 0.239, p = 0.005, n = 104)
and right (τb = 0.28, p = 0.001, n = 99) ventricles. Furthermore, a significant
association was found between clinically known or suspected cardiac amyloidosis
and the vascular score of the right ventricle (τb = 0.181, p = 0.048, n = 99),
though no such relationship was detected in the left ventricle (τb = 0.149, p =
0.09, n = 104).
We did not find any direct correlation
between myocardial fibrosis (either patchy fibrosis or isolated scars) and the
vessel scores of the left (τb = 0.061, p = 0.5, n = 104) or right (τb = −0.038,
p = 0.66, n = 104) ventricle. However, in the subgroup analysis, considering
only patients without stenotic coronary sclerosis (defined here as no coronary
artery showing >50% lumen constriction, as defined in a previous study from
our group [34]), we observed a significant correlation between myocardial
fibrosis and vessel scores in the right ventricle (τb = 0.333, p = 0.04, n = 34;
left ventricle: τb = 0.295, p = 0.06, n = 35).
Correlation to cause of death
The main cause of death was cardiovascular
(50%, n = 52) followed by infectious and inflammatory causes (25%, see appendix
table S4). Cardiac amyloidosis was directly involved in the cause of death in
29 patients (27.9%). Direct cardiac amyloidosis involvement in cause of death
correlated with the interstitial myocardial scores of both ventricles (left: τb =
0.392, p <0.001, n = 104; right: τb = 0.334, p <0.001, n = 99) but not the vascular
scores.
No correlation was found between the sex of
the patient (all n = 104) and amyloidosis type (τb = 0.036, p = 0.72), amyloid
distribution (τb = −0.088, p = 0.37), affected compartment (τb = 0.108, p = 0.25),
clinical diagnosis (τb = 0.184, p = 0.06) or cause of death (τb = 0.014, p = 0.88).
Discussion
This autopsy study was designed to
determine the prevalence and morphological characteristics of cardiac
amyloidosis. Our findings revealed a prevalence of 5.3% for cardiac amyloidosis
in a cohort of 1992 autopsies, with 94.2% of cases classified as amyloid
transthyretin-cardiac amyloidosis (ATTR-CA) and the remaining 5.8% as amyloid
light chain-cardiac amyloidosis (AL-CA). Patients diagnosed with AL-CA were
significantly younger and more frequently exhibited systemic amyloidosis compared
to those with ATTR-CA. Notably, we did not identify any cases of serum amyloid
A-cardiac amyloidosis (AA-CA) in our cohort. This contrasts with a recent study
from Japan, which reported a high prevalence of ATTR-CA (54.2%) alongside a
substantial prevalence of AA-CA (24.4%), while AL-CA accounted for 6.1% of
cases, with the remaining cases being classified as equivocal [28]. These variations
may suggest geographical differences in the prevalence of cardiac amyloidosis
subtypes. In fact, extensive research from Europe and the USA indicates that
ATTR and AL are the predominant forms of cardiac amyloidosis, whereas cardiac
involvement in AA amyloidosis is exceedingly rare [1, 2, 24, 35, 36]. Conversely,
in AA amyloidosis, the kidneys are the most frequently and severely affected organ
[37].
Our data highlight that 94% of cardiac
amyloidosis cases were diagnosed only at autopsy, reinforcing the notion that cardiac
amyloidosis remains a clinically underdiagnosed condition in clinical practice [38,
39]. ATTR-CA, in particular, is more prone to clinical underdiagnosis, likely
due to its higher prevalence in elderly patients [1, 38]. Clinicians may have
greater awareness of AL-CA, given its recognised association with multiple
myeloma and monoclonal gammopathy of undetermined significance [1]. Notably, even
though there was no significant correlation between sex and diagnosis status,
in our cohort most patients with ante mortem diagnosis or clinical suspicion of cardiac
amyloidosis were male (8 male, i.e. 12.9% of all males vs 1 female, i.e. 2.4%
of all females). This sex disparity may further highlight the diagnostic gap
and the necessity for increased clinical vigilance, especially in female
patients.
The routine sampling of heart tissue of the
left and right ventricle at autopsy enabled us to perform a detailed analysis
of the cardiac amyloid distribution. We developed a semi-quantitative scoring
system to correlate amyloid deposits with clinical findings, revealing that the
clinical likelihood of amyloid detection prior to death was significantly
associated with interstitial and vascular amyloid burden in the right ventricle.
In contrast, there was only a weak correlation with left ventricular interstitial
amyloid and no correlation with left ventricular vascular amyloid. Our findings
are in line with current research in the field of multimodality cardiac imaging
that underscores the diagnostic and prognostic significance of right
ventricular involvement in cardiac amyloidosis. Most recently, Datar et al.
demonstrated that 18F-florbetapir PET/CT can detect early right ventricular
amyloid deposition, correlating with dysfunction and major adverse cardiac
events [40]. Similarly, echocardiographic right ventricular strain [41, 42] and
CMR-derived right ventricular strain [43] provides a valuable diagnostic marker
for cardiac amyloidosis.
While the European Society of Cardiology
(ESC) guidelines on cardiomyopathies recommend screening for cardiac
amyloidosis in patients with a left ventricular wall thickness of ≥12 mm [3]
and at least one additional red flag, our results suggest a higher sensitivity
of right ventricular alterations in predicting cardiac amyloidosis. However, it
should be acknowledged that right ventricular assessment is inherently more
challenging in echocardiography compared to left ventricular assessment and
that the left ventricular wall thickness threshold of ≥12mm was chosen to
increase the sensitivity for detecting cardiac amyloidosis, compared to the
higher threshold of ≥14 mm used in other guidelines such as those from the AHA [44].
Nevertheless, it does not imply that the right ventricle is less affected or
that there are no cases of cardiac amyloidosis with a left ventricular wall
thickness of <12mm [45]. Given the growing recognition of right ventricular
amyloid burden in cardiac amyloidosis, our study adds to the expanding body of
evidence highlighting its diagnostic and prognostic value.
Notably, a high vascular amyloid score in
both the left and right ventricles was significantly associated with systemic
amyloidosis. This finding may suggest that detecting vascular amyloid in a
heart biopsy could warrant further screening for systemic amyloidosis. However,
in the clinical setting, the distinction between vascular and interstitial
amyloid deposition is not emphasised at the moment, which represents an
important novel aspect of our study. Given that vascular amyloid deposition may
contribute to systemic dysfunction, it is notable that profound vascular
dysfunction was recently demonstrated in patients with amyloidosis, including
at the retinal level, which could be a consequence of vascular amyloid
accumulation [46]. Moreover, since systemic amyloidosis is generally presumed
and actively investigated – with approximately 95% of ATTR-CA cases diagnosed
via non-biopsy methods such as technetium scintigraphy – and endomyocardial
biopsy is now rarely performed, these results should be interpreted with
caution, particularly given the limited number of cases with systemic
amyloidosis (n = 6). Larger studies are necessary to determine whether vascular
amyloid detection in biopsy specimens holds additional diagnostic value for
systemic amyloidosis. Interestingly, another finding was the significant
association between myocardial fibrosis and vascular amyloidosis in the right
ventricle, which, to the best of our knowledge, has not been previously
reported in the literature. However, this result is based on a small subgroup
analysis that included only patients without significant coronary stenosis. The
size of this group is too limited to allow for definitive interpretations or
further statistical analyses, and the observed association may be incidental or
influenced by biases inherent to an autopsy-based cohort. In this context,
larger studies are needed to further investigate and clarify this phenomenon.
The
current ESC
Guidelines for the management of cardiomyopathies recommend red flags to prompt amyloidosis
screening [3]. This
includes clinical, echocardiography, ECG, CMR and other categories for
initiating early amyloidosis screening. In our study, most patients were diagnosed
ante mortem with hypertensive heart disease (40%), heart failure with a left
ventricular ejection fraction (LVEF) below 50%, and atrial fibrillation
(45.5%). Additionally, common findings in our patient cohort included aortic
valve stenosis (29.1%) and AV conduction disease (27.3%). However, no
statistically significant differences were observed between ATTR-CA and AL-CA
in ante mortem cardiological findings. It is important to note that the number
of AL-CA patients in our study was limited (n=6), which may restrict the
ability to detect significant differences in ante mortem findings. In contrast,
extracardiac findings demonstrated clearer distinctions between the two
subtypes. Macroglossia (p = 0.0004), monoclonal gammopathy of undetermined
significance (p = 0.0004) and haemorrhagic strokes (p = 0.02) were significantly more
frequent in AL-CA than in ATTR-CA, aligning with findings from multiple studies in
the literature [1–4,
47]. Notably, macroglossia is a well-recognised feature exclusive to AL-CA and
is not observed in ATTR-CA, further reinforcing its diagnostic significance in
distinguishing between the two subtypes.
Furthermore, consistent with prior research
[48], patients with AL-CA in our cohort were significantly younger and more
frequently exhibited systemic amyloidosis than those with ATTR-CA did. These
findings highlight key clinical distinctions between the two subtypes, emphasising
the importance of extracardiac manifestations in differentiating AL-CA from
ATTR-CA and underscoring the need for comprehensive diagnostic evaluation.
Further studies are necessary to assess the clinical
relevance of our findings, particularly for cardiac imaging. It remains unclear
how many cases of undiagnosed cardiac amyloidosis could have been detected
prior to death in a clinical setting using current European guideline-based screening
criteria [3]. Additionally, the significance of small amyloid deposits on symptomatology
and life expectancy is uncertain. The process of ageing is associated with
alterations in protein homeostasis, which in turn leads to an increased prevalence
of protein misfolding [49–51]. This complicates determining whether the mere
presence of amyloid deposits, particularly in instances with a low amyloid
burden, has the potential to significantly affect cardiac health. However,
sporadic ATTR-CA has been previously linked to an increased risk of sudden cardiac
death even in the early disease stages [52]. Similarly, a recent study on
forensic autopsies from Australia over a 20-year period (2003–2022) revealed
that cardiac amyloidosis was a contributing factor in 11 deaths, with three being
the primary cause of death [53]. Likewise, AL-CA has been implicated in sudden cardiac
death [54].
Our study has limitations inherent to
retrospective, single-centre autopsy research, including selection bias and a
declining autopsy rate [55]. Despite routine sampling of three left myocardial
regions (anterior wall, posterior wall and septum) and one right myocardial region
in each patient, this approach may not fully represent cardiac amyloidosis
distribution of the entire heart. The absence of sinoatrial node and conduction
tissue sampling prevents conclusions regarding conduction system involvement.
Limited clinical data for some patients also constrained the analysis.
Additionally, most cases were diagnosed postmortem, precluding genetic testing
for hereditary ATTR amyloidosis.
Finally, it is important to consider the
autopsy rate of our institution, which can be calculated as the ratio of adult
autopsies to total adult deaths during the study period, yielding a value of
12.6%. This relatively low rate indicates that the cases included in the study
represent only a small and specific subset of the overall population – primarily
hospitalised patients, often with multiple comorbidities. It is also worth
noting that the selection criteria for determining which deceased patients
undergo autopsy are not clearly defined. Decisions are made on a case-by-case
basis, primarily relying on individual clinical judgement and the request of
the attending physician. Typically, autopsies are performed in cases of
unexplained or unexpected death, complex clinical scenarios or uncertain
diagnoses requiring postmortem confirmation. Consequently, these factors
introduce a potential selection bias, limiting the generalisability of the findings
to the broader population. In this regard, another potential source of bias is
the absence of a control or survivor group, which limits the ability to draw
definitive conclusions about risk factors for cardiac amyloidosis and may
render such discussions largely speculative.
An additional point to consider is that our
study spans a 10-year period (2014–2023) during which clinical awareness and
the incentive to diagnose cardiac amyloidosis have substantially increased due
to the recent availability and reimbursement of disease-modifying therapies.
Therefore, our findings may underestimate current diagnostic rates and reflect
clinical practices that have evolved over the course of the study period.
In
conclusion, our study provides valuable epidemiological insights into cardiac
amyloidosis, highlighting its underdiagnosis. Further
investigation is necessary to determine the clinical implications of our
findings and their impact on patient management.
Data sharing statement
The data underlying this study are of a sensitive
nature and therefore will not be made publicly available on open data
repositories. However, deidentified data may be made available upon reasonable
request, subject to approval by the corresponding author and the Director of
the Department of Autopsy of our institution.
Dr
Umberto Maccio, MD, MSc, MBA
Department
of Pathology and Molecular Pathology
University
Hospital Zürich
Schmelzbergstrasse
12
CH-8091
Zürich
umberto.maccio[at]usz.ch
References
1. Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and
treatment of cardiac amyloidosis: a position statement of the ESC Working Group on
Myocardial and Pericardial Diseases. Eur Heart J. 2021 Apr;42(16):1554–68. doi: https://doi.org/10.1093/eurheartj/ehab072
2. Bloom MW, Gorevic PD. Cardiac Amyloidosis. Ann Intern Med. 2023 Mar;176(3):ITC33–48.
doi: https://doi.org/10.7326/AITC202303210
3. Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, et
al.; ESC Scientific Document Group. 2023 ESC Guidelines for the management of cardiomyopathies.
Eur Heart J. 2023 Oct;44(37):3503–626. doi: https://doi.org/10.1093/eurheartj/ehad194
4. Buxbaum JN, Eisenberg DS, Fändrich M, McPhail ED, Merlini G, Saraiva MJ, et al. Amyloid
nomenclature 2024: update, novel proteins, and recommendations by the International
Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2024 Dec;31(4):249–56.
doi: https://doi.org/10.1080/13506129.2024.2405948
5. Ruberg FL, Maurer MS. Cardiac Amyloidosis Due to Transthyretin Protein: A Review.
JAMA. 2024 Mar;331(9):778–91. doi: https://doi.org/10.1001/jama.2024.0442
6. Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis. Clin Med (Lond). 2018 Apr;18 Suppl
2:s30–5. doi: https://doi.org/10.7861/clinmedicine.18-2-s30
7. Brouwers S, Heimgartner R, Laptseva N, Aguzzi A, Ehl NF, Fehr T, et al. Historic characteristics
and mortality of patients in the Swiss Amyloidosis Registry. Swiss Med Wkly. 2024 Feb;154(2):3485.
doi: https://doi.org/10.57187/s.3485
8. Palladini G, Kastritis E, Maurer MS, Zonder J, Minnema MC, Wechalekar AD, et al. Daratumumab
plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results
of ANDROMEDA. Blood. 2020 Jul;136(1):71–80. doi: https://doi.org/10.1182/blood.2019004460
9. Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, et al. Natural History
of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging
System. J Am Coll Cardiol. 2016 Sep;68(10):1014–20. doi: https://doi.org/10.1016/j.jacc.2016.06.033
10. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016 Jun;387(10038):2641–54.
doi: https://doi.org/10.1016/S0140-6736(15)01274-X
11. Wang S, Peng W, Pang M, Mao L, Peng D, Yu B, et al. Clinical Profile and Prognosis
of Hereditary Transthyretin Amyloid Cardiomyopathy: A Single-Center Study in South
China. Front Cardiovasc Med. 2022 Jun;9:900313. doi: https://doi.org/10.3389/fcvm.2022.900313
12. Conceição I, Damy T, Romero M, Galán L, Attarian S, Luigetti M, et al. Early diagnosis
of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene
mutations. Amyloid. 2019 Mar;26(1):3–9. doi: https://doi.org/10.1080/13506129.2018.1556156
13. Schwotzer R, Flammer AJ, Gerull S, Pabst T, Arosio P, Averaimo M, et al. Expert recommendation
from the Swiss Amyloidosis Network (SAN) for systemic AL-amyloidosis. Swiss Med Wkly.
2020 Dec;150(4950):w20364. doi: https://doi.org/10.4414/smw.2020.20364
14. Castaño A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, et al. Unveiling
transthyretin cardiac amyloidosis and its predictors among elderly patients with severe
aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017 Oct;38(38):2879–87.
doi: https://doi.org/10.1093/eurheartj/ehx350
15. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy:
JACC State-of-the-Art Review. J Am Coll Cardiol. 2019 Jun;73(22):2872–91. doi: https://doi.org/10.1016/j.jacc.2019.04.003
16. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection
fraction. Nat Rev Cardiol. 2017 Oct;14(10):591–602. doi: https://doi.org/10.1038/nrcardio.2017.65
17. AbouEzzeddine OF, Davies DR, Scott CG, Fayyaz AU, Askew JW, McKie PM, et al. Prevalence
of Transthyretin Amyloid Cardiomyopathy in Heart Failure With Preserved Ejection Fraction.
JAMA Cardiol. 2021 Nov;6(11):1267–74. doi: https://doi.org/10.1001/jamacardio.2021.3070
18. Clerc OF, Datar Y, Cuddy SA, Bianchi G, Taylor A, Benz DC, et al. Prognostic Value
of Left Ventricular 18F-Florbetapir Uptake in Systemic Light-Chain Amyloidosis. JACC
Cardiovasc Imaging. 2024 Aug;17(8):911–22. doi: https://doi.org/10.1016/j.jcmg.2024.05.002
19. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic
staging system for light chain amyloidosis incorporating cardiac biomarkers and serum
free light chain measurements. J Clin Oncol. 2012 Mar;30(9):989–95. doi: https://doi.org/10.1200/JCO.2011.38.5724
20. Palladini G, Milani P. Diagnosis and Treatment of AL Amyloidosis. Drugs. 2023 Feb;83(3):203–16.
doi: https://doi.org/10.1007/s40265-022-01830-z
21. Lachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, et al. Natural
history and outcome in systemic AA amyloidosis. N Engl J Med. 2007 Jun;356(23):2361–71.
doi: https://doi.org/10.1056/NEJMoa070265
22. Maccio U, Meier CA, Reinehr M, Ruschitzka F, Schüpbach R, Moch H, et al. Clinically
Undiagnosed Diseases in Autopsies: Frequency and Risk Factors. Arch Pathol Lab Med.
2025 Jan;149(1):60–6. doi: https://doi.org/10.5858/arpa.2023-0429-OA
23. Bajwa F, O’Connor R, Ananthasubramaniam K. Epidemiology and clinical manifestations
of cardiac amyloidosis. Heart Fail Rev. 2022 Sep;27(5):1471–84. doi: https://doi.org/10.1007/s10741-021-10162-1
24. Aimo A, Merlo M, Porcari A, Georgiopoulos G, Pagura L, Vergaro G, et al. Redefining
the epidemiology of cardiac amyloidosis. A systematic review and meta-analysis of
screening studies. Eur J Heart Fail. 2022 Dec;24(12):2342–51. doi: https://doi.org/10.1002/ejhf.2532
25. Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile
systemic amyloidosis affects 25% of the very aged and associates with genetic variation
in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232–9.
doi: https://doi.org/10.1080/07853890701842988
26. Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular
amyloid deposition in patients with heart failure and preserved ejection fraction.
JACC Heart Fail. 2014 Apr;2(2):113–22. doi: https://doi.org/10.1016/j.jchf.2013.11.004
27. Porcari A, Bussani R, Merlo M, Varrà GG, Pagura L, Rozze D, et al. Incidence and Characterization
of Concealed Cardiac Amyloidosis Among Unselected Elderly Patients Undergoing Post-mortem
Examination. Front Cardiovasc Med. 2021 Nov;8:749523. doi: https://doi.org/10.3389/fcvm.2021.749523
28. Tateishi Y, Yamada Y, Katsuki M, Nagata T, Yamamoto H, Kohashi K, et al. Pathological
review of cardiac amyloidosis using autopsy cases in a single Japanese institution.
Pathol Res Pract. 2021 Nov;227:153635. doi: https://doi.org/10.1016/j.prp.2021.153635
29. Ueda M, Sekijima Y, Koike H, Yamashita T, Yoshinaga T, Ishii T, et al. Monitoring
of asymptomatic family members at risk of hereditary transthyretin amyloidosis for
early intervention with disease-modifying therapies. J Neurol Sci. 2020 Jul;414:116813.
doi: https://doi.org/10.1016/j.jns.2020.116813
30. Maccio U, Wicki A, Ruschitzka F, Beuschlein F, Wolleb S, Varga Z, et al. Frequency
and Consequences of Immune Checkpoint Inhibitor-Associated Inflammatory Changes in
Different Organs: An Autopsy Study Over 13 -Years. Mod Pathol. 2025 Apr;38(4):100683.
doi: https://doi.org/10.1016/j.modpat.2024.100683
31. Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc
Pathol. 2015;24(6):343–50. doi: https://doi.org/10.1016/j.carpath.2015.07.008
32. Linke RP. On typing amyloidosis using immunohistochemistry. Detailled illustrations,
review and a note on mass spectrometry. Prog Histochem Cytochem. 2012 Aug;47(2):61–132.
doi: https://doi.org/10.1016/j.proghi.2012.03.001
33. Puth MT, Neuhäuser M, Ruxton GD. Effective use of Spearman’s and Kendall’s correlation
coefficients for association between two measured traits. Anim Behav. 2015;102:77–84.
doi: https://doi.org/10.1016/j.anbehav.2015.01.010
34. Maccio U, Meier CA, Reinehr M, Ruschitzka F, Schupbach R, Moch H, et al. Clinically
Undiagnosed Diseases in Autopsies: Frequency and Risk Factors. Arch Pathol Lab Med.
2024.
35. Fontana M, Ćorović A, Scully P, Moon JC. Myocardial Amyloidosis: The Exemplar Interstitial
Disease. JACC Cardiovasc Imaging. 2019 Nov;12(11 Pt 2):2345–56. doi: https://doi.org/10.1016/j.jcmg.2019.06.023
36. de Marneffe N, Dulgheru R, Ancion A, Moonen M, Lancellotti P. Cardiac amyloidosis:
a review of the literature. Acta Cardiol. 2022 Oct;77(8):683–92. doi: https://doi.org/10.1080/00015385.2021.1992990
37. Mirioglu S, Uludag O, Hurdogan O, Kumru G, Berke I, Doumas SA, et al. AA Amyloidosis:
A Contemporary View. Curr Rheumatol Rep. 2024 Jul;26(7):248–59. doi: https://doi.org/10.1007/s11926-024-01147-8
38. Yun S, Palladini G, Anderson LJ, Cariou E, Wang R, Angeli FS, et al. International
prevalence of transthyretin amyloid cardiomyopathy in high-risk patients with heart
failure and preserved or mildly reduced ejection fraction. Amyloid. 2024 Dec;31(4):291–301.
doi: https://doi.org/10.1080/13506129.2024.2398446
39. Rubin J, Maurer MS. Cardiac Amyloidosis: Overlooked, Underappreciated, and Treatable.
Annu Rev Med. 2020 Jan;71(1):203–19. doi: https://doi.org/10.1146/annurev-med-052918-020140
40. Datar Y, Clerc OF, Cuddy SA, Kim S, Taylor A, Neri JC, et al. Quantification of right
ventricular amyloid burden with 18F-florbetapir positron emission tomography/computed
tomography and its association with right ventricular dysfunction and outcomes in
light-chain amyloidosis. Eur Heart J Cardiovasc Imaging. 2024 Apr;25(5):687–97. doi: https://doi.org/10.1093/ehjci/jead350
41. Ozbay B, Satyavolu BS, Rearick C, Soman P, Katz WE, Sezer A, et al. Right Ventricular
Strain Improves the Echocardiographic Diagnosis and Risk Stratification of Transthyretin
Cardiac Amyloidosis Among Other Phenotypes of Left Ventricular Hypertrophy. J Am Soc
Echocardiogr. 2024 Oct;37(10):947–59. doi: https://doi.org/10.1016/j.echo.2024.06.006
42. Istratoaie S, Bourg C, Lee KC, Marut B, Antonelli J, L’official G, et al. Right ventricular
free wall strain predicts transthyretin amyloidosis prognosis as well as biomarker-based
staging systems. Eur Heart J Cardiovasc Imaging. 2025 Jan;26(2):239–48. doi: https://doi.org/10.1093/ehjci/jeae242
43. Eckstein J, Körperich H, Weise Valdés E, Sciacca V, Paluszkiewicz L, Burchert W, et
al. CMR-based right ventricular strain analysis in cardiac amyloidosis and its potential
as a supportive diagnostic feature. Int J Cardiol Heart Vasc. 2022 Dec;44:101167.
doi: https://doi.org/10.1016/j.ijcha.2022.101167
44. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al.; ACC/AHA
Joint Committee Members. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure:
A Report of the American College of Cardiology/American Heart Association Joint Committee
on Clinical Practice Guidelines. Circulation. 2022 May;145(18):e895–1032. doi: https://doi.org/10.1161/CIR.0000000000001063
45. Nagy D, Révész K, Peskó G, Varga G, Horváth L, Farkas P, et al. Cardiac Amyloidosis
with Normal Wall Thickness: Prevalence, Clinical Characteristics and Outcome in a
Retrospective Analysis. Biomedicines. 2022 Jul;10(7):1765. doi: https://doi.org/10.3390/biomedicines10071765
46. Zampiccoli E, Barthelmes J, Kreysing L, Nägele MP, Nebunu D, Haider T, et al. Eyes
on amyloidosis: microvascular retinal dysfunction in cardiac amyloidosis. ESC Heart
Fail. 2022 Apr;9(2):1186–94. doi: https://doi.org/10.1002/ehf2.13792
47. Chen D, Zhang C, Parikh N, Merkler AE, Navi BB, Fink ME, et al. Association Between
Systemic Amyloidosis and Intracranial Hemorrhage. Stroke. 2022 Mar;53(3):e92–3. doi: https://doi.org/10.1161/STROKEAHA.121.038451
48. Clerc OF, Cuddy SA, Jerosch-Herold M, Benz DC, Katznelson E, Canseco Neri J, et al. Myocardial
Characteristics, Cardiac Structure, and Cardiac Function in Systemic Light-Chain Amyloidosis.
JACC Cardiovasc Imaging. 2024 Nov;17(11):1271–86. doi: https://doi.org/10.1016/j.jcmg.2024.05.004
49. Stroo E, Koopman M, Nollen EA, Mata-Cabana A. Cellular Regulation of Amyloid Formation
in Aging and Disease. Front Neurosci. 2017 Feb;11:64. doi: https://doi.org/10.3389/fnins.2017.00064
50. Cannata’ A, Merlo M, Artico J, Gentile P, Camparini L, Cristallini J, et al. Cardiovascular
aging: the unveiled enigma from bench to bedside. J Cardiovasc Med (Hagerstown). 2018 Oct;19(10):517–26.
doi: https://doi.org/10.2459/JCM.0000000000000694
51. Pras A, Nollen EA. Regulation of Age-Related Protein Toxicity. Front Cell Dev Biol.
2021 Mar;9:637084. doi: https://doi.org/10.3389/fcell.2021.637084
52. Ichimata S, Hata Y, Hirono K, Yamaguchi Y, Nishida N. Clinicopathological features
of clinically undiagnosed sporadic transthyretin cardiac amyloidosis: a forensic autopsy-based
series. Amyloid. 2021 Jun;28(2):125–33. doi: https://doi.org/10.1080/13506129.2021.1882979
53. Tan L, Byard RW. Cardiac amyloid deposition and the forensic autopsy - A review and
analysis. J Forensic Leg Med. 2024 Apr;103:102663. doi: https://doi.org/10.1016/j.jflm.2024.102663
54. D’Errico S, Mazzanti A, Baldari B, Maiese A, Frati P, Fineschi V. Sudden death in
lambda light chain AL cardiac amyloidosis: a review of literature and update for clinicians
and pathologists. Int J Clin Exp Pathol. 2020 Jul;13(7):1474–82.
55. Rodewald AK, Bode P, Cathomas G, Moch H. [Clinical autopsies in Switzerland: A status
report]. Pathologe. 2017 Sep;38(5):416–21. doi: https://doi.org/10.1007/s00292-017-0323-8
Appendix
The appendix is available in the pdf version of the article at https://doi.org/10.57187/s.4145.