Genetically engineered T cell and tumour-infiltrating lymphocyte therapies
DOI: https://doi.org/https://doi.org/10.57187/s.4279
Andreas
Holbroabc,
Heinz
Läublibde
a Division of Haematology, University
Hospital Basel, Basel, Switzerland
b Innovation Focus Cell Therapies,
University Hospital Basel, Basel, Switzerland
c Regional Blood Transfusion Service,
Swiss Red Cross, Basel, Switzerland
d Department of Biomedicine,
University of Basel, Basel, Switzerland
e Division of Oncology, University
Hospital Basel, Basel, Switzerland
Summary
Haemato-oncology has made significant
progress in recent years, particularly through the development of innovative
immunotherapeutic approaches such as CAR T cell (chimeric antigen receptor T
cell) and tumour-infiltrating lymphocyte therapies. Both methods use the
patient’s own immune system to treat cancer, but in different ways. CAR T cell
therapy is a form of immunotherapy in which the patient’s own T cells are
genetically modified. CAR T cell therapies have proven to be particularly
effective in haematological B-cell neoplasms, such as B-cell acute
lymphoblastic leukaemia (B-ALL) and B-cell lymphomas, as well as in multiple
myeloma. Tumour-infiltrating lymphocyte therapy, on the other hand, exploits the
natural ability of T cells to recognise tumour-associated antigens of tumour
cells with the T cell receptor. Tumour tissue is taken from the patient then tumour-infiltrating
lymphocytes are isolated from it. These tumour-infiltrating lymphocytes are expanded
ex vivo to increase their number and activity. This review discusses the
principles of these innovative therapies. Both therapies represent significant
advances in personalised cancer treatment and offer new hope for our cancer
patients.
Abbreviations
- B-ALL:
-
acute B-cell lymphoblastic leukaemia
- CAR:
-
chimeric antigen receptor
- DLBCL:
-
diffuse large B-cell lymphoma
- scFv:
-
single-chain variable fragment
- TIL:
-
tumour-infiltrating lymphocyte
Introduction
Cancer immunotherapy has significantly
improved the outcome of patients. In particular, the introduction of immune
checkpoint inhibitors has transformed the treatment of patients with solid
cancers [1, 2]. However, only a limited number of patients benefit from immune
checkpoint inhibition [3]. Cell therapies including genetically engineered T
cells expressing a chimeric antigen receptor (CAR) have been used to treat
cancer patients [4]. While such CAR T cell therapies have been successfully
used mainly to treat patients with haematological B cell neoplasia, naturally
occurring tumour-infiltrating lymphocytes (TILs) have been used for the
treatment of solid cancers resistant to immune checkpoint inhibitors [5]. Here,
we give an overview of these two emerging cell therapy approaches (figure 1)
and an outlook on current developments.
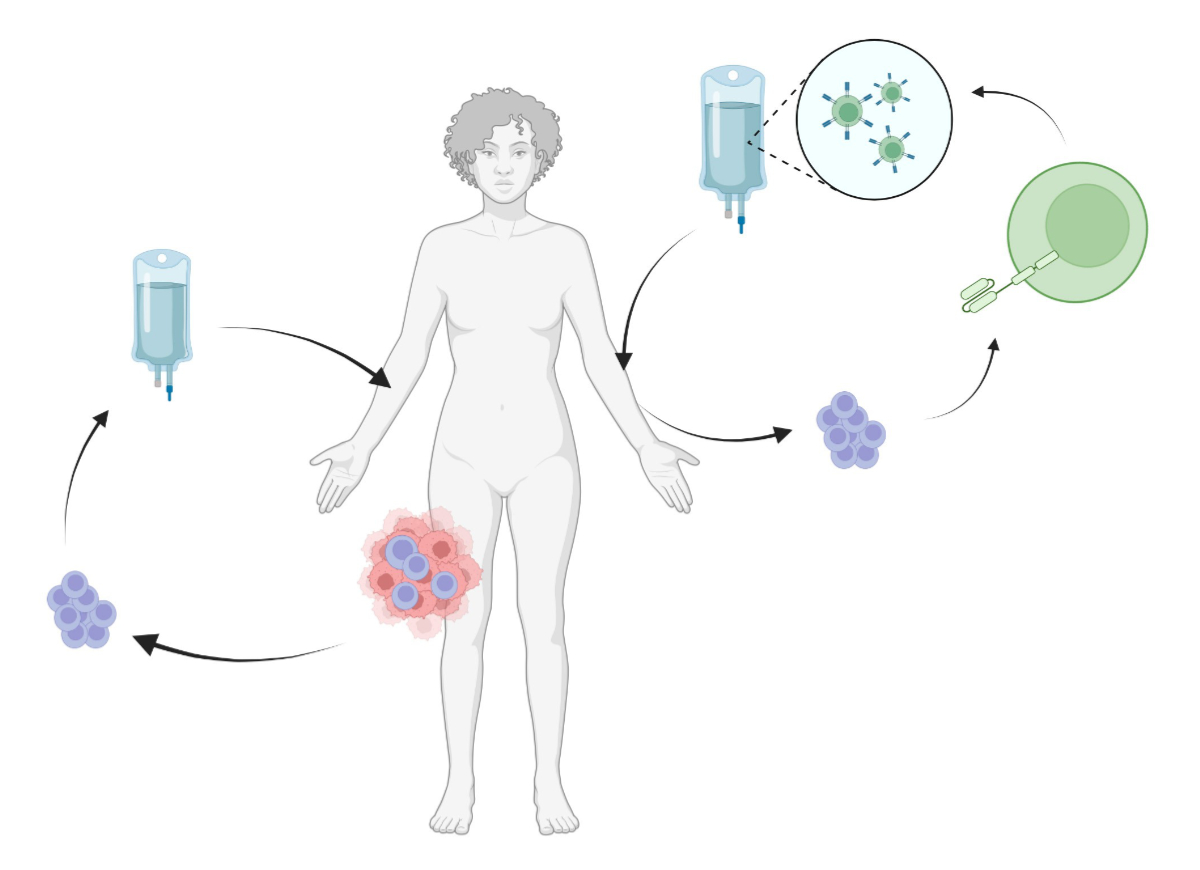
Figure 1Illustration of tumour-infiltrating
lymphocyte (TIL) therapy (left) and CAR T cell therapy. In tumour-infiltrating
lymphocyte therapy, T cells are isolated from the tumour and expanded to >50
× 109 cells with IL-2, CD3 stimulation and allogeneic
feeders. After lymphodepletion with cyclophosphamide and fludarabine, the
CD3-positive T cells are administered to the patient together with IL-2. CAR T
cell therapy uses T cells obtained by apheresis. The T cells are then
genetically manipulated (usually with a lenti- or retroviral vector) so that
they stably express a CAR. They are then returned to the patient who has
undergone lymphodepletion.
CAR T cell therapy
Cellular cancer therapies have been used to
treat cancer since the introduction of allogeneic stem cell transplantation by
Don Thomas in the 1970s [6]. In the late 1980s, T cells were genetically
modified for the first time, equipping them with
a synthetic T cell receptor containing an intracellular activation domain, the
CD3ζ of the T cell receptor, and an extracellular binding domain, usually a single-chain
variable fragment (scFv) of an antibody directed against a surface molecule on
the cancer cell [7] (figure 2). These so-called chimeric antigen receptors
(CARs) were further developed, showing a good effect in mouse models but also
in patients with B-cell neoplasia [4]. Further development led to the so-called
second-generation CARs, which also contained a co-stimulatory component in the
intracellular domain [4]. The intracellular domain of CD28 or 4-1BB (CD137) was
primarily used for this purpose (figure 2). This enabled not only recognition
of the target antigen but also proliferation and persistence of these cells in
vivo. More recent developments include other co-stimulatory domains or
synthetic proteins that lead to an increase in specificity or efficiency as
well as the possibility of better control of their proliferation. In contrast
to antibodies targeting a tumour surface antigen such as the anti-CD20 antibody
rituximab, CAR T cells can persist and lead to long-term control by the immune
system.
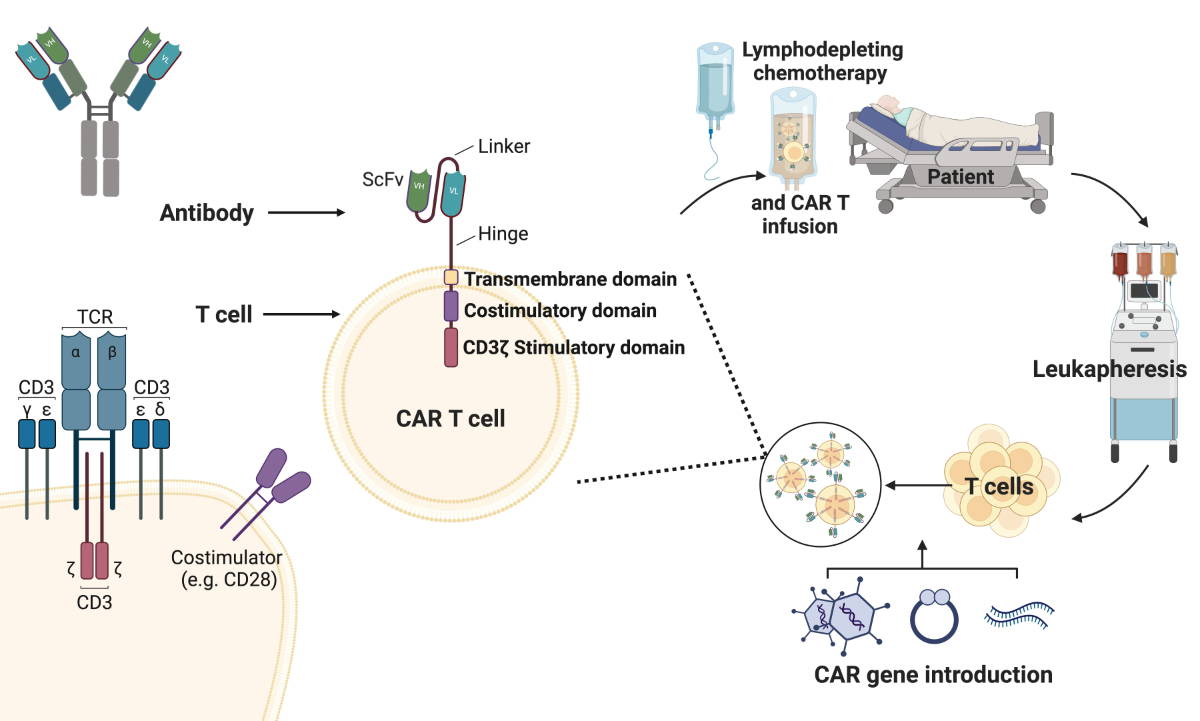
Figure 2Demonstration of the genetic
modification of T cells during CAR T cell production. Chimeric antigen
receptors (CARs) are synthetic proteins consisting of an intracellular part
with different signalling domains and an antigen-binding domain that is
commonly derived from an antibody and expressed as a short-chain variable
fragment (ScFv). CAR constructs are delivered to T cells after a leukapheresis
and retransfused in the lymphodepleted patient. TCR: T cell receptor. Created
with BioRender.com.
Haematological diseases
The treatment of B cell malignancies with
cellular immunotherapies has become the standard of care for several
indications [4, 8]. Treatment is currently approved when initial treatment has
failed or in refractory cases (table 1). In particular, CD19-positive B cell
malignancies have been successfully treated with CD19-specific CAR T cells [4,
8]. CAR T cell therapy was able to achieve durable remissions in patients after
multiple prior lines of therapy and in some cases highly
chemotherapy-refractory disease. Recently, several studies have also shown an
advantage of CAR T cell therapy in patients with relapsed/refractory diffuse
large B-cell lymphoma compared to the standard of care with chemotherapy
followed by autologous stem cell transplantation (diffuse large B-cell lymphoma)
[4, 8]. For example, in the ZUMA-7 study, axicabtagene-ciloleucel (Axi-cel) was
tested in 359 patients compared to standard treatment [9]. In addition to the
CD19-targeted CAR T cell therapies, CAR T cells against the BCMA antigen have
also been established for the treatment of multiple myeloma [10, 11]. Other
targets on myeloma cells such as SLAMF-7 or G protein-coupled receptor, class
C, group 5, member D (GPRC5D) are currently being investigated [12].
Table 1EMA-approved CAR T therapies for B
cell malignancies and multiple myeloma (as of September 2024).
| Product |
Kymriah |
Yescarta |
Tecartus |
Breyanzi |
Abecma |
Carvykti |
| Active substance |
Tisagenlecleucel |
Axicabtagene-ciloleucel |
Brexucabtagene autoleucel |
Lisocabtagene maraleucel |
Idecabtagene-vicleucel |
Ciltacabtagenum autoleucelum |
| Manufacturer |
Novartis |
Kyte/Gilead |
Kyte/Gilead |
BMS/Celgene |
BMS/Celgene |
Janssen |
| Approval (EMA) |
2018 |
2018 |
2020 |
2022 |
2021 |
2022 |
| Target |
CD19 |
CD19 |
CD19 |
CD19 |
BCMA |
BCMA |
| Costimulatory signal |
4-1BB |
CD28 |
CD28 |
4-1BB |
4-1BB |
4-1BB |
| Indication(s) |
r/r B-ALL (age ≤25, 3rd line); r/r DLBC (3rd line); r/r FL (3rd line) |
r/r DLBCL, HGBCL (2nd line*); PMBCL (3rd
line); r/r FL (4th line) |
r/r MCL (3rd line; previous lines included
BTK inhibitor); r/r B-ALL (age ≥26, 3rd
line) |
r/r DLBCL, PMBCL, HGBCL (2nd line*); FL grade 3B (2nd line*) |
r/r MM (3rd line**) |
r/r MM (2nd line***) |
Solid cancers
Several targets for solid tumours are
currently under investigation. Early trials for HER2-positive cancers were
terminated due to severe toxicity on target tissues outside the tumour (so-called
on-target/off-tumour effects) [4, 8]. Major hurdles for CAR T cell therapy in
solid malignancies include defining the correct tumour antigen with specific,
high-level expression in the tumour, difficulties in trafficking T cells into
the tumour microenvironment and depletion of CAR T cells due to an
immunosuppressive tumour microenvironment [4, 8]. Nevertheless, some successful
studies have already been conducted. For example, a promising response to
claudin 18.2-targeted CAR T cells has been shown in patients with advanced
gastric or pancreatic cancer [13]. An interesting approach was pursued by
Mackensen and colleagues. In addition to the use of claudin 6-directed CAR T
cells, a combination with an RNA vaccine, which induces additional activation
of the CAR T cells with enhancement of a memory function, was pursued. The
study showed promising results in gastrointestinal and gynaecological
malignancies [14]. In recent months, several studies on the treatment of
glioblastoma with CAR T cells have also been published [15, 16]. One by Bagley
et al. used intrathecally administered CAR T cells able to recognise two
antigens on glioblastoma cells, namely EGFR and IL13Ralpha2 [16]. The other by
Marcela Maus’ group used locally administered CAR T cells that recognise both the
tumour-specific
variant of EGFRvIII and the wild-type variant of EGFR. Promising results were
also achieved with anti-GD2 CAR T cells in paediatric gliomas, with complete
remissions being induced [17]. So far, however, the long-term results are very
sobering; they show that the principle also works in solid tumours but that
further work is needed to achieve longer-term tumour control or eradication.
Recently, the first genetically engineered cell product for solid tumours has
been approved by the FDA; however it is not a CAR T cell therapy but a T cell
receptor T cell therapy targeting the tumour antigen MAGE-A4. Afamitresgene
autoleucel was successfully tested in HLA-A*02:01-, 02-, 03- and 06-positive
patients with metastatic synovial sarcoma [18].
Various additional measures can potentially
increase the efficiency of CAR T cells against solid tumours. For example, to
increase the specificity of the CAR construct, genetic systems have been
developed that can integrate two or more tumour-transmitted signals [4, 8]. The
synNOTCH system, for example, uses the Notch signalling system to mediate a
two-step activation of CAR T cells [19]. The binding of a first antigen induces
the expression of the activating CAR, which leads to the activation of the
immune cell. This two-step CAR T cell activation can significantly increase the
specificity of such a CAR T cell product. The local release of cytokines that
promote anti-tumour immunity could improve the activation and efficacy of CAR T
cells [8]. For example, secretion of IL-12 could enhance CD19-targeted CAR T
cell therapy [20].
CAR T cell therapy for patients with autoimmune
disease
Recently, several reports have been
published describing a role for CAR T cell therapy in patients with autoimmune
disease [21–27]. For example, patients with treatment-refractory systemic lupus
erythematosus (SLE) treated with CD19-directed CAR T cells experienced significant
improvement [21–24]. Also, CD19-targeted CAR T cell therapies were used to
treat patients with treatment-refractory anti-synthetase syndrome [25, 26, 28]
or patients with multiple sclerosis and myasthenia gravis [27, 29].
Side effects of CAR T cell therapy
CAR T cell therapy is considered a
promising cancer treatment but is associated with various side effects, some of
which can be life-threatening [30]. One of the most common side effects is
cytokine release syndrome (CRS), which can cause fever, low blood pressure and
organ dysfunction. In addition, immune effector cell-associated neurotoxicity
syndrome (ICANS) can occur, a reversible but potentially life-threatening
complication that can manifest with confusion, seizures and other neurological
symptoms [31]. In phase III trials of second-line therapy for patients with
diffuse large B-cell lymphoma, second-generation CAR T cells showed that up to
90% of patients experienced cytokine release syndrome and 60% neurological side
effects [32]. However, most of these side effects were not severe and were
manageable. Mild cytokine release syndrome is treated with supportive measures
such as antipyretics and hydration. More severe cytokine release syndrome
requires the use of IL-6R blocking antibodies and immunosuppressants such as
corticosteroids [33]. In severe cases, patients may need to be transferred to
the ICU for circulatory monitoring and possibly oxygen or respiratory support.
While IL-6 blockade is often effective in cytokine release syndrome, it does
not help in immune effector cell-associated neurotoxicity syndrome and may even
worsen the clinical picture. In addition to supportive measures,
corticosteroids are used here, possibly in combination with anakinra.
Improvements in CAR design could potentially reduce these sometimes
life-threatening side effects. It is known that CARs with a CD28 co-stimulatory
domain (e.g. axi-cel) proliferate faster and release higher cytokine
concentrations, which can lead to earlier and more-severe cytokine release
syndrome and immune effector cell-associated neurotoxicity syndrome compared to
4-1BB-containing CARs (e.g. tisa-cel) [31]. Initial studies suggest that
changes in the signalling domain of the CD3ζ chain may improve the side effect
profile. Medium-term complications include cytopenias, infections and disease
relapse. In the latter case, the therapeutic options are very limited and the
prognosis is very poor. An increased risk of secondary malignancies has been
reported but further studies, in particular to better understand the
association, are warranted. Further challenges are the numerous resources and
logistics that such a therapy requires. As a result, the costs are also very
high.
Tumour-infiltrating lymphocytes
The use of cellular therapy with
tumour-infiltrating lymphocytes has been practiced for several decades, but
only in specialised clinics [34–36]. The first patients were treated in the
late 1980s [35]. In tumour-infiltrating lymphocyte therapy, T cells are
isolated from a sample of the primary tumour or a metastasis [37]. By using
IL-2, T cells are activated and multiplied. The treatment works in some
patients because tumour-specific T cell clones are present in the resected
lesion. These tumour-specific T cells have a T cell receptor that recognises an
antigen expressed by the tumour. After lymphodepletion with cyclophosphamide
and fludarabine, the tumour-infiltrating lymphocyte product is administered
together with IL-2 [37]. Recently, a randomised phase III study conducted in
the Netherlands and Denmark in patients with metastatic melanoma was published [5].
Here it was shown that after failure of a standard immunotherapy with immune
checkpoint inhibition against PD-1, patients responded better to tumour-infiltrating
lymphocyte therapy than to a second-line immune checkpoint inhibition with
antibodies directed against CTLA-4. In addition, a commercial product, lifileucel,
was recently approved by the FDA in the USA for the treatment of melanoma
patients. We have recently conducted the BaseTIL study, in which we treated 9
patients with advanced and heavily pre-treated melanoma [38]. In addition to
melanoma patients, in principle all patients with T cells containing T cell
receptors that recognise a tumour antigen (often so-called neoantigens, which
arise through new mutations in cancer cells) can be treated. Patients with
non-small cell lung cancer (NSCLC) have also been successfully treated with tumour-infiltrating
lymphocytes [39]. Other immunogenic tumour entities such as cervical carcinoma
can also potentially be treated with tumour-infiltrating lymphocyte therapy. We
have recently opened a trial for patients with NSCLC at the University Hospital
in Basel (NCT06455917).
Tumour-infiltrating lymphocyte therapies
can of course also be improved. For example, the T cells that recognise tumour
antigens can be separated from the other T cells in the tumour. Thus,
potentially much larger numbers of T cells attacking the tumour can be isolated
and amplified [37, 40]. In order to obtain a more functional phenotype of the T
cells, other cytokines or stimulating antibodies can also be added for in vitro
expansion [40]. Tumour-infiltrating lymphocyte therapy can also be combined
with other substances. For example, tumour-infiltrating lymphocyte therapy has
already been combined with immune checkpoint inhibitors in various studies. In
some cases, a good response to this combination therapy was observed in
patients with NSCLC [41]. Other forms of IL-2 can also be used. There are newer
IL-2 preparations that can specifically stimulate the cytotoxic T cells rather
than the regulatory T cells [42]. We are currently conducting the BaseTIL-03M
study in Basel, which is testing a combination of such a new IL-2 preparation
together with tumour-infiltrating lymphocyte therapy in melanoma patients
(NCT05869539).
Side effects of tumour-infiltrating lymphocyte
therapy
Cytopenias following lymphodepleting
chemotherapy and thus infections and bleeding complications are the most common
side effects of tumour-infiltrating lymphocyte therapy [5]. Interleukin-2
therapy can also cause cytokine release syndrome, depending on the dose and intensity
of IL-2 administration. In addition to cytokine release syndrome, IL-2
treatment can also lead to vascular leak syndrome, sometimes with pulmonary oedema
[37]. It is therefore important to consider the use of vasoactive substances at
an early stage. As tumour-infiltrating lymphocyte therapy is a classic
immunotherapy, immune-mediated side effects with various organ toxicities can
also occur, as with immune checkpoint inhibitor therapy [40]. These often have
to be treated with steroids, depending on which organs are involved.
Outlook
Emerging cellular therapies, such as CAR T
cell therapy and tumour-infiltrating lymphocyte therapy, have significantly
improved prognosis for certain patients. CAR T cell therapies are now routinely
used for haematological cancers, primarily B-cell lymphomas, and efforts are
underway to expand CAR T cell treatments to additional tumour types. Moreover,
CAR T cell therapies may soon offer new treatment options for selected patients
with refractory autoimmune diseases and a multitude of studies are ongoing [14].
Tumour-infiltrating lymphocyte therapy also holds promise for inducing
long-term remissions in patients with immunogenic tumours. With the recent
approval of a commercial tumour-infiltrating lymphocyte product for treating
melanoma in the United States, this treatment has become accessible to a wider
patient population. Trials are currently evaluating its effectiveness for other
tumour types, and advancements in expanding tumour-specific T cells are
expected to enhance its efficacy.
The availability of these cellular
therapies will place new demands on treatment centres, as more patients are
expected to seek these specialised options. While the costs of these therapies,
including associated treatments and potential complications, remain high,
future logistical improvements may help manage expenses. For example, some centres
are already producing tumour-infiltrating lymphocyte therapies in their own
cleanroom facilities, which may reduce production costs over time.
Additionally, certain preparatory and therapeutic steps may transition to
outpatient settings, making these therapies more accessible.
Andreas
Holbro, MD
Division of
Haematology
University
Hospital Basel
Petersgraben
4
CH-4031
Basel
andreas.holbro[at]usb.ch
and
Heinz
Läubli
Division of
Oncology
University
Hospital Basel
Petersgraben
4
CH-4031 Basel
heinz.laeubli[at]unibas.ch
References
1. Bicak M, Cimen Bozkus C, Bhardwaj N. Checkpoint therapy in cancer treatment: progress,
challenges, and future directions. J Clin Invest. 2024 Sep;134(18):e184846. doi: https://doi.org/10.1172/JCI184846
2. Sharma P, Goswami S, Raychaudhuri D, Siddiqui BA, Singh P, Nagarajan A, et al. Immune
checkpoint therapy-current perspectives and future directions. Cell. 2023 Apr;186(8):1652–69.
doi: https://doi.org/10.1016/j.cell.2023.03.006
3. Haslam A, Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are
Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open.
2019 May;2(5):e192535. doi: https://doi.org/10.1001/jamanetworkopen.2019.2535
4. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018 Jul;379(1):64–73.
10.1056/NEJMra1706169
5. Rohaan MW, Borch TH, van den Berg JH, Met Ö, Kessels R, Geukes Foppen MH, et al. Tumor-Infiltrating
Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N Engl J Med. 2022 Dec;387(23):2113–25.
doi: https://doi.org/10.1056/NEJMoa2210233
6. Thomas ED. Bone marrow transplantation from the personal viewpoint. Int J Hematol.
2005 Feb;81(2):89–93. doi: https://doi.org/10.1532/IJH97.04197
7. Pinthus JH, Waks T, Malina V, Kaufman-Francis K, Harmelin A, Aizenberg I, et al. Adoptive
immunotherapy of prostate cancer bone lesions using redirected effector lymphocytes.
J Clin Invest. 2004 Dec;114(12):1774–81. doi: https://doi.org/10.1172/JCI200422284
8. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next
generation. Nature. 2023 Feb;614(7949):635–48. doi: https://doi.org/10.1038/s41586-023-05707-3
9. Westin JR, Oluwole OO, Kersten MJ, Miklos DB, Perales MA, Ghobadi A, et al.; ZUMA-7
Investigators; Kite Members. Survival with Axicabtagene Ciloleucel in Large B-Cell
Lymphoma. N Engl J Med. 2023 Jul;389(2):148–57. doi: https://doi.org/10.1056/NEJMoa2301665
10. Rodriguez-Otero P, Ailawadhi S, Arnulf B, Patel K, Cavo M, Nooka AK, et al. Ide-cel
or Standard Regimens in Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2023 Mar;388(11):1002–14.
doi: https://doi.org/10.1056/NEJMoa2213614
11. Sheykhhasan M, Ahmadieh-Yazdi A, Vicidomini R, Poondla N, Tanzadehpanah H, Dirbaziyan A,
et al. CAR T therapies in multiple myeloma: unleashing the future. Cancer Gene Ther.
2024 May;31(5):667–86. doi: https://doi.org/10.1038/s41417-024-00750-2
12. Mailankody S, Devlin SM, Landa J, Nath K, Diamonte C, Carstens EJ, et al. GPRC5D-Targeted
CAR T Cells for Myeloma. N Engl J Med. 2022 Sep;387(13):1196–206. doi: https://doi.org/10.1056/NEJMoa2209900
13. Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in
gastrointestinal cancers: phase 1 trial interim results. Nat Med. 2022 Jun;28(6):1189–98.
doi: https://doi.org/10.1038/s41591-022-01800-8
14. Mackensen A, Haanen JB, Koenecke C, Alsdorf W, Wagner-Drouet E, Borchmann P, et al. CLDN6-specific
CAR-T cells plus amplifying RNA vaccine in relapsed or refractory solid tumors: the
phase 1 BNT211-01 trial. Nat Med. 2023 Nov;29(11):2844–53. doi: https://doi.org/10.1038/s41591-023-02612-0
15. Choi BD, Gerstner ER, Frigault MJ, Leick MB, Mount CW, Balaj L, et al. Intraventricular
CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N Engl J Med. 2024 Apr;390(14):1290–8.
doi: https://doi.org/10.1056/NEJMoa2314390
16. Bagley SJ, Logun M, Fraietta JA, Wang X, Desai AS, Bagley LJ, et al. Intrathecal bivalent
CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim
results. Nat Med. 2024 May;30(5):1320–9. doi: https://doi.org/10.1038/s41591-024-02893-z
17. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR
T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022 Mar;603(7903):934–41.
doi: https://doi.org/10.1038/s41586-022-04489-4
18. D’Angelo SP, Araujo DM, Abdul Razak AR, Agulnik M, Attia S, Blay JY, et al. Afamitresgene
autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1):
an international, open-label, phase 2 trial. Lancet. 2024 Apr;403(10435):1460–71.
doi: https://doi.org/10.1016/S0140-6736(24)00319-2
19. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR
T cells overcome challenges of specificity, heterogeneity, and persistence in treating
glioblastoma. Sci Transl Med. 2021 Apr;13(591):eabe7378. doi: https://doi.org/10.1126/scitranslmed.abe7378
20. Smole A, Benton A, Poussin MA, Eiva MA, Mezzanotte C, Camisa B, et al. Expression
of inducible factors reprograms CAR-T cells for enhanced function and safety. Cancer
Cell. 2022 Dec;40(12):1470–1487.e7. doi: https://doi.org/10.1016/j.ccell.2022.11.006
21. Schett G, Müller F, Taubmann J, Mackensen A, Wang W, Furie RA, et al. Advancements
and challenges in CAR T cell therapy in autoimmune diseases. Nat Rev Rheumatol. 2024 Sep;20(9):531–44.
doi: https://doi.org/10.1038/s41584-024-01139-z
22. Mougiakakos D, Krönke G, Völkl S, Kretschmann S, Aigner M, Kharboutli S, et al. CD19-Targeted
CAR T Cells in Refractory Systemic Lupus Erythematosus. N Engl J Med. 2021 Aug;385(6):567–9.
doi: https://doi.org/10.1056/NEJMc2107725
23. Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19
CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. 2022 Oct;28(10):2124–32.
doi: https://doi.org/10.1038/s41591-022-02017-5
24. Müller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Völkl S, et al. CD19 CAR T-Cell
Therapy in Autoimmune Disease - A Case Series with Follow-up. N Engl J Med. 2024 Feb;390(8):687–700.
doi: https://doi.org/10.1056/NEJMoa2308917
25. Taubmann J, Knitza J, Müller F, Völkl S, Aigner M, Kleyer A, et al. Rescue therapy
of antisynthetase syndrome with CD19-targeted CAR-T cells after failure of several
B-cell depleting antibodies. Rheumatology (Oxford). 2024 Jan;63(1):e12–4. doi: https://doi.org/10.1093/rheumatology/kead330
26. Pecher AC, Hensen L, Klein R, Schairer R, Lutz K, Atar D, et al. CD19-Targeting CAR
T Cells for Myositis and Interstitial Lung Disease Associated With Antisynthetase
Syndrome. JAMA. 2023 Jun;329(24):2154–62. doi: https://doi.org/10.1001/jama.2023.8753
27. Granit V, Benatar M, Kurtoglu M, Miljković MD, Chahin N, Sahagian G, et al.; MG-001
Study Team. Safety and clinical activity of autologous RNA chimeric antigen receptor
T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label,
non-randomised phase 1b/2a study. Lancet Neurol. 2023 Jul;22(7):578–90. doi: https://doi.org/10.1016/S1474-4422(23)00194-1
28. Müller F, Boeltz S, Knitza J, Aigner M, Völkl S, Kharboutli S, et al. CD19-targeted
CAR T cells in refractory antisynthetase syndrome. Lancet. 2023 Mar;401(10379):815–8.
doi: https://doi.org/10.1016/S0140-6736(23)00023-5
29. Fischbach F, Richter J, Pfeffer LK, Fehse B, Berger SC, Reinhardt S, et al. CD19-targeted
chimeric antigen receptor T cell therapy in two patients with multiple sclerosis.
Med (N Y). 2024 Jun;5(6):550–558.e2. doi: https://doi.org/10.1016/j.medj.2024.03.002
30. Brudno JN, Kochenderfer JN. Current understanding and management of CAR T cell-associated
toxicities. Nat Rev Clin Oncol. 2024 Jul;21(7):501–21. doi: https://doi.org/10.1038/s41571-024-00903-0
31. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated
neurotoxicity in cancer immunotherapy. Nat Rev Immunol. 2022 Feb;22(2):85–96. doi: https://doi.org/10.1038/s41577-021-00547-6
32. Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, et al.; All
ZUMA-7 Investigators and Contributing Kite Members. Axicabtagene Ciloleucel as Second-Line
Therapy for Large B-Cell Lymphoma. N Engl J Med. 2022 Feb;386(7):640–54. doi: https://doi.org/10.1056/NEJMoa2116133
33. Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy.
Blood. 2023 May;141(20):2430–42.
34. Klobuch S, Seijkens TT, Schumacher TN, Haanen JB. Tumour-infiltrating lymphocyte therapy
for patients with advanced-stage melanoma. Nat Rev Clin Oncol. 2024 Mar;21(3):173–84.
doi: https://doi.org/10.1038/s41571-023-00848-w
35. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use
of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients
with metastatic melanoma. A preliminary report. N Engl J Med. 1988 Dec;319(25):1676–80.
doi: https://doi.org/10.1056/NEJM198812223192527
36. Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized,
Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer
of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J Clin Oncol.
2016 Jul;34(20):2389–97. doi: https://doi.org/10.1200/JCO.2016.66.7220
37. Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy
for cancer. Immunol Rev. 2014 Jan;257(1):56–71. doi: https://doi.org/10.1111/imr.12132
38. König D, Sandholzer MT, Uzun S, Zingg A, Ritschard R, Thut H, et al. Melanoma clonal
heterogeneity leads to secondary resistance after adoptive cell therapy with tumor-infiltrating
lymphocytes. Cancer Immunol Res. 2024 Jul;12(7):814–21. doi: https://doi.org/10.1158/2326-6066.CIR-23-0757
39. Schoenfeld AJ, Lee SM, Doger de Spéville B, Gettinger SN, Häfliger S, Sukari A, et
al. Lifileucel, an Autologous Tumor-Infiltrating Lymphocyte Monotherapy, in Patients
with Advanced Non-Small Cell Lung Cancer Resistant to Immune Checkpoint Inhibitors.
Cancer Discov. 2024 Aug;14(8):1389–402. doi: https://doi.org/10.1158/2159-8290.CD-23-1334
40. Rohaan MW, van den Berg JH, Kvistborg P, Haanen JB. Adoptive transfer of tumor-infiltrating
lymphocytes in melanoma: a viable treatment option. J Immunother Cancer. 2018 Oct;6(1):102.
doi: https://doi.org/10.1186/s40425-018-0391-1
41. Creelan BC, Wang C, Teer JK, Toloza EM, Yao J, Kim S, et al. Tumor-infiltrating lymphocyte
treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat Med.
2021 Aug;27(8):1410–8. doi: https://doi.org/10.1038/s41591-021-01462-y
42. Joerger M, Calvo E, Laubli H, Lopez J, Alonso G, Corral de la Fuente E, et al. Phase
1 first-in-human dose-escalation study of ANV419 in patients with relapsed/refractory
advanced solid tumors. J Immunother Cancer. 2023 Nov;11(11):e007784. doi: https://doi.org/10.1136/jitc-2023-007784