Cardiac
amyloidosis
DOI: https://doi.org/https://doi.org/10.57187/s.4186
Natallia Laptsevaab,
Dominik C. Benzabc,
Rahel Schwotzerad,
Andreas J. Flammerab
a Amyloidosis Network Zurich,
University Hospital Zurich, Zurich, Switzerland
b University Heart Center, University
Hospital Zurich, Zurich, Switzerland
c Cardiac Imaging, Department of
Nuclear Medicine, University Hospital of Zurich, Zurich, Switzerland
d Hematology and Oncology,
University Hospital Zurich, Zurich, Switzerland
Summary
Cardiac amyloidosis is a disease
characterised by the accumulation of amyloid protein in the heart tissue. There
are several types of amyloidosis, but the most common types affecting the heart
are ATTR amyloidosis (caused by transthyretin protein) and AL amyloidosis
(caused by abnormal immunoglobulin light chains). Cardiac amyloidosis causes
typical signs and symptoms of heart failure. Diagnosis involves a combination
of imaging tests such as echocardiography and cardiac magnetic resonance
imaging, as well as nuclear imaging scans and tissue biopsies to confirm the
presence of amyloid deposits in the heart. Treatment of cardiac amyloidosis depends
on the type and severity of the disease and includes medications to manage
symptoms as well as treatments targeting the underlying cause of amyloidosis.
Importantly, cardiac amyloidosis is a serious condition requiring specialised
care from a multidisciplinary team including cardiologists and haematologists
as well as other specialists familiar with the management of this rare disease.
This is crucial, as early diagnosis and treatment are important for improving
outcomes.
Introduction
Amyloidosis
is a storage disorder occurring when extracellular deposited amyloid leads to
dysfunction of various organs. Amyloid is formed in the presence of a protein-folding
disease. Currently, more than 30 different precursor proteins are known; however,
the two most common forms are AL (light chain) and ATTR (transthyretin)
amyloidosis.
Despite the
heterogeneity of the precursor proteins, the ultrastructural morphology and
histochemical properties of amyloid fibrils are remarkably similar. They share
a common core structure of antiparallel β-strands perpendicular to the long
axis of the fibril [1]. This extremely abnormal, highly ordered conformation
underlies the characteristic properties of amyloid fibrils, including their
relative stability and resistance to proteolysis, as well as their ability to
bind molecules of Congo red dye, resulting in pathognomonic apple-green
birefringence when viewed under cross-polarised light [2]. Despite the
histological similarity of the amyloid itself, the mechanisms leading to the
formation of the precursor proteins are completely different, and thus the
pathophysiology of the disease and, of course, the options for causal therapy
are completely different.
AL
amyloidosis can occur in any form of B-cell monoclonal dyscrasia, typically in
plasma cell dyscrasia [3]. Instead of producing normal immunoglobulins, plasma
cell or mature B-cell clones produce pathological components (light chains of
monoclonal immunoglobulin), which can aggregate to form amyloid fibrils.
The liver
is the main organ producing transthyretin precursor proteins in ATTR
amyloidosis. Transthyretin is a transport protein for thyroid hormones and
retinol. Transthyretin is a tetramer consisting of four monomers. Normally, the
tetramers dissociate to monomers and aggregate back again [4]. With age
(wild-type ATTR [5]), the progressive dissociation of tetramers into monomers
results in the accumulation of monomers in blood and aggregation into amyloid
fibrils. In rare cases, a mutation in the TTR gene may cause the liver
to produce a pathological transthyretin form (variant ATTR – hereditary ATTR-amyloidosis
[4].
Damage to the organs
arises, on the one hand, from deposition of fibrils into the tissue and, on the
other hand, through direct cytotoxic effects of circulating fibrils and
precursor fibrils (proteotoxicity) [6]. Further, fibrils exhibit organotropy,
tending to deposit in the heart, ligaments and nerves in case of ATTRv,
while AL amyloid deposits mainly into the heart, kidney, gastrointestinal tract
and nervous system.
Prognosis
Prognosis
of untreated AL amyloidosis is extremely poor, particularly if the heart is
affected (mean survival is 6–15 months and 10-year survival rate
is below 5%) [3]. The more the heart is affected, the worse the prognosis [7]; however,
the prognosis of AL amyloidosis has substantially improved with new treatment
options in recent years [8, 9].
The
prognosis of wtATTR amyloidosis is generally better than in AL amyloidosis.
Several scores predict survival. In the most commonly used Gillmore score,
prognosis mainly depends on NT-proBNP values and estimated GFR [10], with the
best prognosis observed when NT-proBNP is below 3000 ng/l and estimated GFR is higher
than 45 ml/min.
Prevalence
While the incidence
and prevalence of AL amyloidosis remain stable (approximately 1–2 cases per 100,000
subjects [11], the incidence and prevalence of ATTR amyloidosis have been increasing
over the past few years [12] and may be around 4–17 cases per 100,000 [12, 13].
This is mainly due to the ageing population, greater awareness and better
diagnostic tools, particularly scintigraphy. Interestingly, ATTR cardiac amyloidosis
is strongly associated with aortic stenosis (15%) [5] – approximately 16% of
patients in whom transfemoral aortic valve implantation (TAVI) was performed
for severe aortic stenosis showed concomitant ATTR cardiac amyloidosis [14]. In
patients with heart failure and preserved ejection fraction (HFpEF), ATTR cardiac
amyloidosis can be found in as many as 13% of the cases [15]. Furthermore,
approximately 25% of patients aged over 80 years who died were found to have
transthyretin deposits in the heart [16].
Clinical
presentation
In cardiac
amyloidosis, signs and symptoms of heart failure are often the first
manifestation, particularly due to volume overload. However, a large subset of
patients present with thoracic complaints such as angina pectoris or orthostasis
and syncope (for typical signs and symptoms, see table 1).
Table 1Typical
findings in amyloidosis.
| Cardiac signs and symptoms |
Shortness of breath (hypervolaemia) |
| Oedema – Volume retention |
| Thoracic pain (microvascular angina, elevated filling
pressures) |
| Orthostatic dysregulation, orthostatic hypotension |
| Syncope |
| Fatigue |
| Palpitations – Arrhythmias |
| Thromboembolism – Stroke |
| Non-cardiac signs and symptoms |
Periorbital purpura (AL amyloidosis) |
| Macroglossia (AL amyloidosis) |
| Skin bruising – periorbital ecchymosis (AL amyloidosis) |
| Bilateral carpal tunnel syndrome |
| Biceps tendon rupture |
| Lumbar spinal stenosis |
| Sensory and motor peripheral neuropathy (vATTR) |
| Weakness |
| GI symptoms (nausea, diarrhoea, weight loss) – vATTR and AL |
| Sexual dysfunction |
| Vitreous opacification, glaucoma (vATTR) |
| ECG findings |
Pseudo Q waves |
| Atrial fibrillation |
| AV conduction disease |
| Widened QRS complex |
| Ventricular premature beats |
| Low voltage (AL) |
| Laboratory findings |
Elevated troponin T and NT-proBNP |
| Impaired kidney function (AL or cardiorenal in ATTR) |
| Proteinuria (AL) |
Interestingly,
patients with ATTR amyloidosis may already be complaining about orthopaedic
manifestations 5–15 years before cardiac amyloidosis becomes symptomatic –
particularly carpal tunnel syndrome, spinal canal stenosis and biceps tendon
rupture. These findings are generally attributed to amyloidosis only after cardiac
amyloidosis is diagnosed [16].
Cardiac
amyloidosis is associated with ECG abnormalities. A typical sign is the pseudo-infarction
pattern, found in up to 83% of patients [17]. While low voltage in the limb
leads has low sensitivity for ATTR amyloidosis, an abnormal voltage-to-mass
ratio occurs in at least 70% of cardiac amyloidosis [18]. In addition, cardiac
amyloidosis is typically associated with conduction disease [19].
Diagnostic work-up
The
presence of typical signs and symptoms of cardiac amyloidosis should prompt further
testing by echocardiography. Inherently, left ventricular wall thickness
represents the diagnostic hallmark of cardiac amyloidosis [20]. The minimal
threshold to screen for cardiac amyloidosis has recently been lowered to 12 mm
by a European consensus statement [20]. Characteristic echocardiographic
findings of cardiac amyloidosis include pleural or pericardial effusion; thickening
of the right ventricle, valves or interatrial septum; a low stroke volume; diastolic
dysfunction; and a paradoxical low-flow low-gradient aortic stenosis (figure 1)
[21]. A reduced longitudinal global strain with apical sparing is another
characteristic feature, which may be sensitive but has limited specificity [22].
If echocardiography has a poor acoustic window or echocardiographic findings
are not suggestive of cardiac amyloidosis, cardiac magnetic resonance imaging
(CMR) may distinguish structural or functional abnormalities better. Typical
findings include increased left and right ventricular mass (or at least wall
thickness), abnormal gadolinium kinetics, diffuse transmural or subendocardial
late gadolinium enhancement, increased T1 mapping or increased extracellular
volume, which has the highest specificity above 40% (figure 1) [23, 24]. Even
though CMR may be highly suggestive, it is not diagnostic of cardiac
amyloidosis.
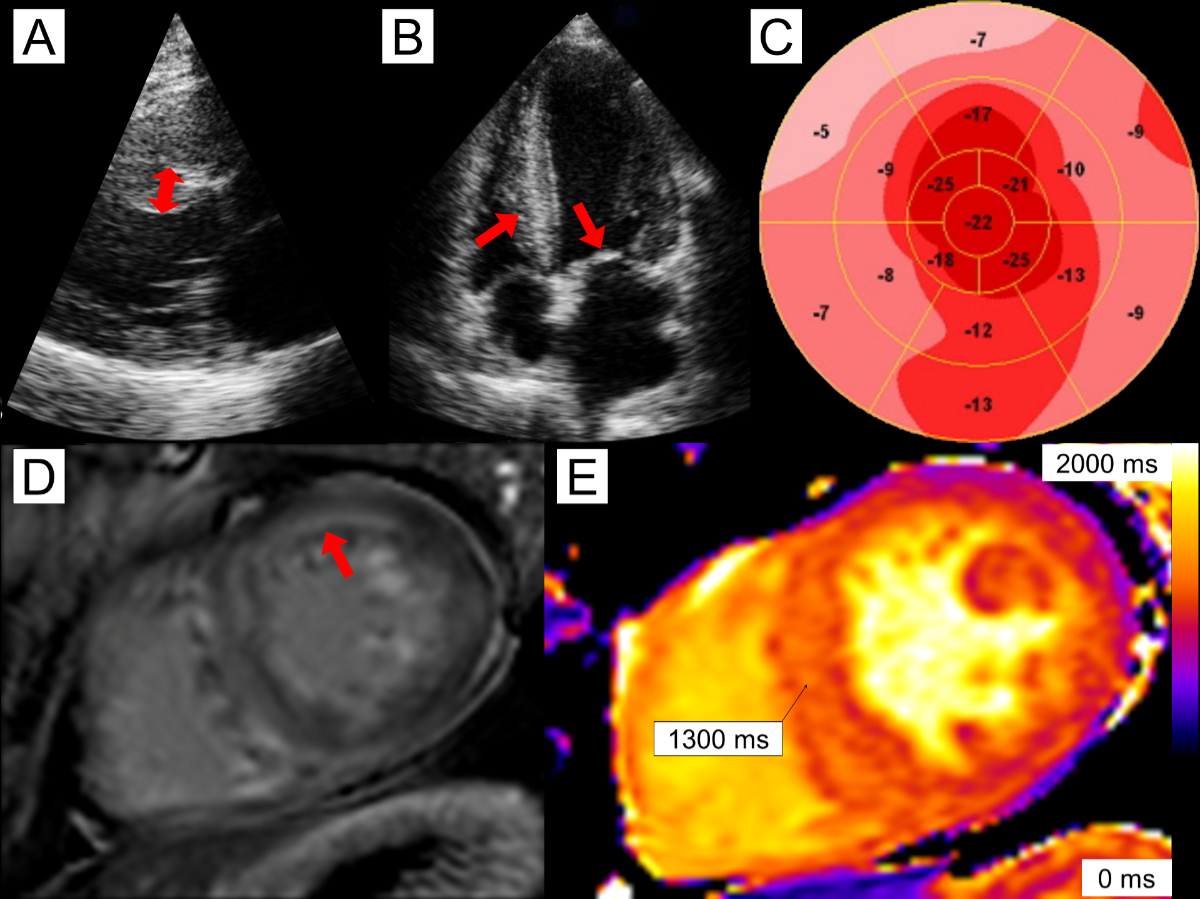
Figure 1A 56-year-old
male with cardiac AL (light chain) amyloidosis. Echocardiography reveals mild asymmetric
left
ventricular (LV) hypertrophy with septal wall thickness of 14 mm (panel A, double-headed arrow) as well as
thickening of the right ventricle (RV) and mitral valve (panel B, arrows). There is reduced global longitudinal strain of
–14.3% and relative apical sparing (panel
C). Cardiac magnetic resonance imaging reveals diffuse, predominantly
subendocardial late gadolinium enhancement (panel
D, arrow). Native T1 times were elevated in the septum, measuring 1300 ms (panel E).
When signs
and symptoms, ECG and echocardiography (or CMR) are suggestive of cardiac
amyloidosis, further testing is indicated to (a) diagnose cardiac amyloidosis
and (b) determine the subtype of cardiac amyloidosis. Until recently, cardiac
amyloidosis was only diagnosed by a positive biopsy, but accumulating
literature supports the notion that cardiac scintigraphy with bone-avid tracers
(DPD, HMDP or PYP) can non-invasively diagnose the disease [20]. However,
certain limitations need to be considered:
- When positive, cardiac scintigraphy
(which is a planar 2D scan) always needs to be complemented by single-photon
emission computed tomography (SPECT) to rule out misinterpretation of blood
pool activity [25].
- Cardiac scintigraphy with bone-avid
radiotracers has a sensitivity over 99% for diagnosing cardiac ATTR amyloid
deposits in cardiac biopsy. The low number of false-negative findings is mainly
due to rare hereditary ATTR forms (i.e. Val30Met and Phe64Leu) [26].
- The specificity of cardiac
scintigraphy with bone-avid radiotracers is 68% because cardiac scintigraphy
detects other non-ATTR forms of cardiac amyloidosis (e.g. cardiac AL
amyloidosis). However, the sensitivity for diagnosing these non-ATTR forms is
much lower than for cardiac ATTR amyloidosis. For example, 61% of patients with
cardiac AL amyloidosis have grade 0 and only 10% have grade 2 or 3 in cardiac
DPD scintigraphy.
Therefore, and as outlined in the
diagnostic algorithm of the Swiss Amyloidosis Network [27], monoclonal
gammopathy needs to be excluded prior to referral to cardiac scintigraphy. This
includes quantification of serum free light chains as well as serum and urine
immunofixation. In an isolated free light chain abnormality (i.e. normal
immunofixation), abnormal kappa/lambda ratio may be explained by kidney
dysfunction, and eGFR-adjusted ratios are used without affecting the specificity
of the test [28].
If
monoclonal gammopathy is absent (figure 2), patients should be referred to
nuclear medicine.
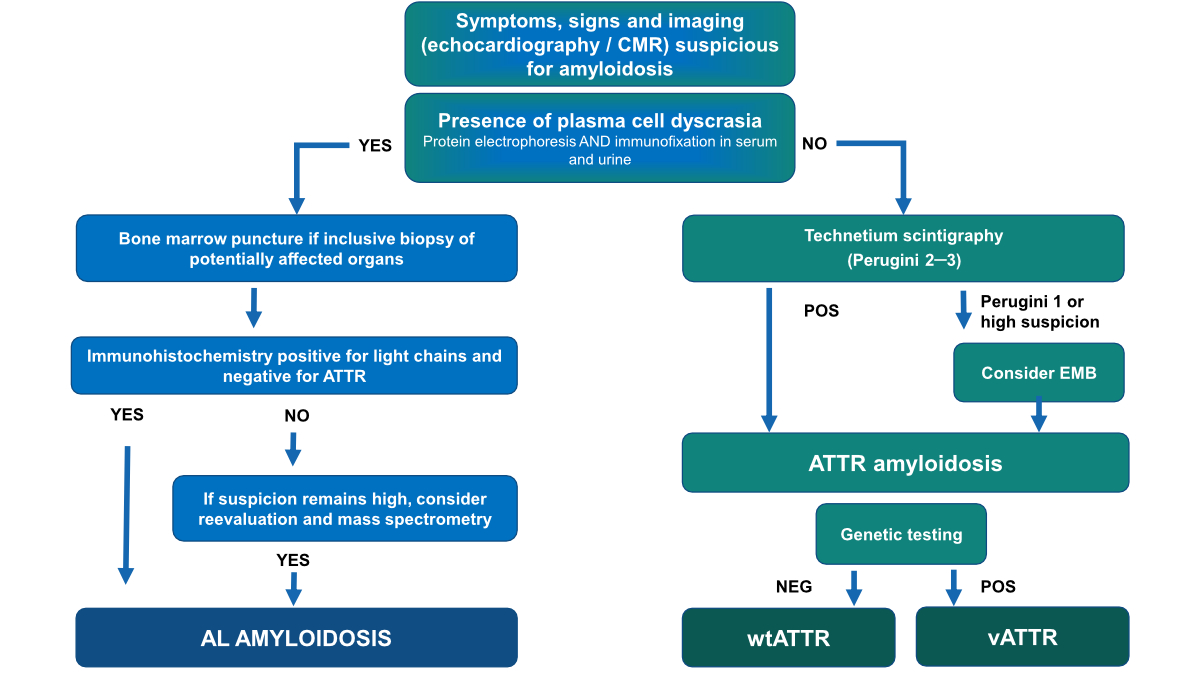
Figure 2Diagnostic algorithm for the diagnosis of cardiac amyloidosis.
When
cardiac scintigraphy/SPECT with bone-avid tracers is positive (i.e. grade 2 or
3), the patient can be diagnosed with cardiac ATTR amyloidosis without biopsy. By
adhering to this diagnostic algorithm, the diagnostic performance can be
summarised as following:
1. After
exclusion of monoclonal gammopathy, cardiac scintigraphy/SPECT with bone-avid
tracers has a specificity of 100% for diagnosing cardiac amyloidosis [26].
2. The
sensitivity of the algorithm is 74% for two reasons:
- About 20% of these elderly patients
suffer from concomitant monoclonal gammopathy of unknown significance (MGUS).
In these patients, endomyocardial or any other organ biopsy is indicated to
differentiate ATTR from AL amyloidosis.
- Some false-negative cases are due to
grade 0 or grade 1 of hereditary or early wild-type forms of ATTR amyloidosis [26].
This highlights the fact that high clinical suspicion of cardiac amyloidosis
should always trigger further testing with CMR or cardiac biopsy, even if
cardiac scintigraphy is negative.
If
monoclonal gammopathy is present (figure 2), patients should be referred to haematology
for further testing including CMR to evaluate cardiac involvement. In case of
plasma cell or mature B-cell dyscrasia, a tissue biopsy must be carried out, usually
of the most affected organ [29]. If cardiac amyloidosis is suspected, a diagnosis
is commonly made with endomyocardial biopsy. However, a biopsy of another
organ, together with typical findings on echocardiography or CMR is valid for
the diagnosis of cardiac amyloidosis [29].
Patients
with plasma cell dyscrasia due to MGUS pose a particular diagnostic challenge,
as this condition is common in elderly patients with ATTR cardiac amyloidosis,
and is significantly higher than in the general population [30, 31].
Tissue
biopsies are evaluated for the presence of amyloid. Typing is generally done
using immunohistochemical staining; however, this technique is challenging and
prone to errors. In certain cases, the biopsy needs to be assessed by a
specialised centre to ascertain the prognosis. Mass spectrometry (MS) proteomic
analysis, the gold standard, can only be performed at very few centres
worldwide.
MS directly identifies the protein subunit in the deposit and the accompanying
universal amyloid proteins. MS can detect unusual or novel types and its
sensitivity and specificity are close to 100% [31].
Currently,
cardiac AL amyloidosis cannot be diagnosed non-invasively, so many patients
undergo endomyocardial biopsy [29]. When AL amyloidosis was detected in an
extracardiac biopsy and echocardiography or CMR are suggestive of cardiac
amyloidosis, AL cardiomyopathy may be diagnosed. More recently, in clinical
trials, amyloid-binding positron emission tomography (PET) radiotracers like 18F-florbetapir
or 124I-evuzamitide have evolved to diagnose cardiac AL amyloidosis
non-invasively [32] and quantify cardiac amyloid burden. In the near future,
this may hold clinical implications for monitoring treatment response [33].
Treatment
Symptomatic
treatment of cardiac amyloidosis
High left
ventricular filling pressure due to severe diastolic dysfunction is the main
clinical problem of patients with cardiac amyloidosis. Therefore, diuretic
treatment, together with patient education to reduce fluid intake, remain the
mainstay of treatment. Diuretic treatment is challenging due to
over-proportional blood pressure decline with an altered pressure/volume
relationship.
Patients
with cardiac amyloidosis mainly present with HFpEF. In these patients,
traditional heart failure treatment with renin-angiotensin-aldosterone inhibition
is not established. A recent large retrospective analysis in more than 2000
ATTR cardiac amyloidosis patients did not show benefit of ACE inhibitors or angiotensin-receptor
blockers on outcome, but a high rate of withdrawal due to side effects [34].
Beta-blockers have been used in cardiac amyloidosis patients; however, low
heart rates should be avoided because cardiac output is solely dependent on
heart rate given that stroke volume is fixed in cardiac amyloidosis. In the
later analysis, beta-blocker therapy showed
benefit in patients with cardiac amyloidosis and an ejection fraction below 40%
(HFrEF) [34]; however, the withdrawal rate due to intolerance remains high. Mineralocorticoid
receptor antagonists, however, showed a better tolerability and lower mortality
and morbidity in these ATTR patients [34].
SGLT-2 inhibitors
are the mainstay of heart failure treatment irrespective of ejection fraction.
There are no signs of harm with SGLT-2 inhibitors in cardiac amyloidosis patients,
in AL and ATTR alike [35]. However, as well as with MRAs more data on outcomes are
needed [36, 37].
Very few
data are available for patients with AL cardiac amyloidosis. In these patients,
the abovementioned drugs are usually not tolerated and may induce severe
symptomatic orthostasis.
Prevention and
treatment of arrhythmias
Arrhythmias
are very common in cardiac amyloidosis. The most common arrhythmia in cardiac
amyloidosis is, by far, atrial fibrillation. Almost all patients develop atrial
fibrillation over time, due to elevated filling pressures and structural
changes in the left atrium. Therefore, regular screening for atrial
fibrillation is important (at 6-month intervals). Stroke risk in cardiac
amyloidosis is very high, particularly in AL cardiac amyloidosis [19]. This is
why oral anticoagulation is mandatory if atrial fibrillation is present. Even
in sinus rhythm, the risk of stroke is increased – this may be due to elevated
left atrial filling pressure and reduced atrial contraction [19]. Therefore,
many groups initiate oral anticoagulation even in the absence of atrial
fibrillation. There are no data on atrial appendage closure and very limited
data on atrial fibrillation ablation in amyloidosis. One study showed a
recurrence rate of almost 90% in the latter [38]. Therefore, we do not
recommend these interventions in cardiac amyloidosis. Rhythm control should be attempted
with electroconversion and amiodarone; however, the success rate is not very
high. Beta-blocker therapy or sometimes amiodarone can be used for rate control;
however, a lenient strategy should be adopted, due to the fixed stroke volume [38].
Digoxin should be used, if at all, only under very careful monitoring [39]. Finally,
as in any case of atrial fibrillation refractory to medical therapy,
atrioventricular nodal ablation and a permanent pacemaker implant can be considered
[40].
Scarring,
fibrosis and amyloid itself may have proarrhythmogenic properties leading to
the emergence of tachyarrhythmias [41]. Although sudden cardiac death (SCD) is
more common in cardiac amyloidosis (particularly AL cardiac amyloidosis) than
in other cardiomyopathies and studies show a high number of appropriate (and
inappropriate) shocks, no study has so far convincingly demonstrated a mortality
benefit with ICD in patients with cardiac amyloidosis [42]. Nevertheless, the
2022 ESC Guidelines for the management of patients with ventricular arrhythmias
advocated for ICD implantation in patients who have haemodynamically
not-tolerated ventricular tachycardias (class IIA, C
recommendation) [43]. Certainly, the decision concerning ICD implantation
should be made in an amyloidosis centre after in-depth discussion with the team
and the patient.
Toxicity of
amyloid fibrils may lead to sinus bradycardia up to sinus arrest and amyloid
deposition may delay impulse propagation within the conduction system [41].
Over time, about 10% of cardiac amyloidosis patients eventually require a
pacemaker. The presence of a first-degree AV block, wide QRS complex (over 120 ms)
and atrial fibrillation indicate the highest risk for future pacemaker
implantation [44], highlighting the need for regular ECG monitoring.
Disease-modifying
treatments
The
treatment of AL cardiac amyloidosis is mainly in the domain of
haemato-oncology, and it varies according to the underlying disease, renal and neurological
parameters, and cardiac involvement. In recent years, treatment options have
increased substantially, and life expectancy increased. Treatment of AL is a
huge topic in itself, but not a focus of this review. Notably, AL patients
should be treated in an amyloidosis centre with an opportunity for an
interdisciplinary approach [45].
For ATTR cardiac
amyloidosis, the only causal treatment currently approved is the tetramer
stabiliser tafamidis. It prevents dissociation of the transthyretin tetramer to
the four monomers, thereby preventing the build-up of amyloid oligomers and
fibrils. Tafamidis was initially developed for the treatment of familial
amyloid polyneuropathy and has been used in this indication for more than 10
years (approved in the EU but not in Switzerland). The “ATTR-act study” [46]
proved the efficacy of tafamidis for the treatment of cardiac amyloidosis.
Mortality and heart failure hospitalisation were significantly lowered compared
to placebo; however, this benefit seems to be achieved after 18 months only.
The effect on quality of life and exercise capacity was seen much faster.
Currently, tafamidis 61 mg/d is approved for the treatment of cardiac
amyloidosis in Switzerland (for limitations, see table 2). Recently, the
effect of tetramer stabilisation on outcome has been confirmed with acoramidis,
a tetramer stabiliser similar to tafamidis [47].
Table 2Limitations from the “Spezialitätenliste” for the treatment of cardiac amyloidosis in Switzerland with tafamidis
61 mg/d.
The drug is only reimbursed by the health insurance when all of the following are
present:
| Established
diagnosis with typical imaging findings along with exclusion of AL amyloidosis
and a positive Tc scintigraphy (Perugini 2–3) or
histological proof of ATTR |
| NYHA
class I or II |
| At least
one prior hospitalisation for heart failure and/or an episode of a
symptomatic documented heart failure |
| NT-proBNP
> 600 ng/l |
| Able to
walk more than 100 m in a 6-minute walk test |
| Glomerular
filtration rate > 25 ml/min/1.73 m2 |
| Life
expectancy of at least 2 years |
| No prior
liver or heart transplantation, no “mechanical assist devices” |
| Must not be combined with other specific
drugs for the treatment of TTR amyloidosis (e.g. patisiran, inotersen) |
| Cardiology
centre, included in the list of the Swiss Federal Office of Public Health |
Importantly,
drug development in cardiac amyloidosis is very dynamic and several new
compounds show promising results for the treatment of cardiac amyloidosis. RNA therapeutics
(siRNA and antisense oligonucleotides) can suppress the production of TTR in
the liver effectively, thus eliminating the protein responsible for TTR
amyloidosis.
For
hereditary ATTR amyloidosis, three substances are already on the market for the
treatment of amyloid polyneuropathy. Patisiran and vutisiran are small
interfering RNAs (si-RNA) binding to transthyretin messenger RNA (mRNA) to
mediate its premature degradation, thereby inhibiting its translation into transthyretin
protein. Similarly, the antisense oligonucleotides inotersen and eplontersen
inhibit the production of transthyretin.
The Apollo-A
(patisiran) [48], Helios-A (vutisiran) [49] and Neuro-TTR (inotersen) [50], NEURO-TTRansform
(eplontersen) [51] studies showed that the substances slowed or even halted
the progression of neuropathy in these patients. The efficacy of these
substances in cardiac amyloidosis patients with wild-type and hereditaryamyloidosis
is promising. In the
recently published Apollo-B trial, administration of patisiran over 12 months
resulted in preserved functional capacity in ATTR cardiac amyloidosis [52]. Very recently,
the Helios-B study demonstrated
significant reduction of death and cardiovascular events with vutrisiran in
patients with ATTR-CM [53]. Of note, RNA therapeutics
for hereditary amyloidosis currently can only be prescribed in one of the
amyloidosis centres at the university hospitals of Lausanne (CHUV) or Zurich
(USZ).
Furthermore,
with CRISPR-Cas9, gene silencing has been achieved in patients with hereditary
amyloidosis and first-in-man data look very promising. This could be one of the
first gene therapies applied to humans [54]. Recently, Fontana et al. described anti-ATTR
antibodies in patients who had recovered from ATTR amyloidosis, highlighting
the possibility for reversibility [55]. Further, an exciting phase I study proved
the concept that amyloid can be cleared from the tissue via an antibody-mediated
phagocytotic inflammatory reaction [56]. After infusion of the antibody,
cardiac tracer update on scintigraphy and extracellular volume on cardiac MRI were
reduced after 12 months. An ongoing phase 3 trial is evaluating this promising
new treatment option.
Conclusion
Overall, amyloidosis has gained a lot
of attention in the last couple of years. This is mainly based on much better
diagnostic tools and treatment options. However, the diagnosis and treatment
remain challenging, and misdiagnosis may pose a danger to the patient, and
potential treatment may be unintentionally withheld. Further, amyloidosis
remains a multiorgan disease and a multidisciplinary approach, particularly in
vATTR and AL amyloidosis, is crucial. Thus, amyloidosis networks, in which the
relevant disciplines work together, are critically important and a nationwide
strategy is helpful in improving quality of care [27, 57, 58].
Key points
- Cardiac
amyloidosis is characterised by the deposition of abnormal proteins called
amyloid in heart tissue. These deposits impair the structure and function of
the heart, leading to various symptoms and complications.
- There are
two main types of amyloid proteins causing cardiac amyloidosis, with the most
common types being ATTR and AL amyloidosis.
- Symptoms
of cardiac amyloidosis vary, but mostly include unexplained heart failure
symptoms in a patient with a thickened septum on echocardiography. Other red flags
include hypotension in a previously hypertensive patient, syncope, unexplained
stroke, bilateral carpal tunnel syndrome, ECG abnormalities (pseudo-Q waves)
and others.
- Diagnosis
is made by typical imaging findings with echocardiography or CMR, together with
a positive technetium scintigraphy in the absence of plasma cell dyscrasia
(ATTR) or via tissue biopsy (AL or ATTR).
- The treatment
goals for cardiac amyloidosis are to manage symptoms and to give medication
targeting the underlying cause of amyloidosis.
- Management
of cardiac amyloidosis requires a multidisciplinary approach, involving
cardiologists, haematologists, nephrologists and other specialists. Close
monitoring and coordination of care are essential to ensure high-quality
treatment.
Acknowledgments
Author
contributions: NL, DB, RS
and AJF contributed substantially to the conception and design of the work and
drafted the work or reviewed it critically for important intellectual content.
All authors approved the final version. All authors are accountable for the
work done.
Prof. Dr. Andreas
Flammer, FESC, FHFA
Head Heart Failure
and Transplantation, Co-Head Amyloidosis-Network Zurich
Cardiology
University Hospital of Zurich
Raemistrasse 100
CH-8091 Zurich
andreas.flammer[at]usz.ch
References
1. Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure
of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997 Oct;273(3):729–39.
doi: https://doi.org/10.1006/jmbi.1997.1348
2. Röcken C, Sletten K. Amyloid in surgical pathology. Virchows Arch. 2003 Jul;443(1):3–16.
doi: https://doi.org/10.1007/s00428-003-0834-y
3. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features
in 474 cases. Semin Hematol. 1995 Jan;32(1):45–59.
4. Jeyashekar NS, Sadana A, Vo-Dinh T. Protein amyloidose misfolding: mechanisms, detection,
and pathological implications. Methods Mol Biol. 2005;300:417–35. doi: https://doi.org/10.1385/1-59259-858-7:417
5. Nitsche C, Scully PR, Patel KP, Kammerlander AA, Koschutnik M, Dona C, et al. Prevalence
and Outcomes of Concomitant Aortic Stenosis and Cardiac Amyloidosis. J Am Coll Cardiol.
2021 Jan;77(2):128–39. doi: https://doi.org/10.1016/j.jacc.2020.11.006
6. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A
Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016 Sep;68(12):1323–41. doi: https://doi.org/10.1016/j.jacc.2016.06.053
7. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic
staging system for light chain amyloidosis incorporating cardiac biomarkers and serum
free light chain measurements. J Clin Oncol. 2012 Mar;30(9):989–95. doi: https://doi.org/10.1200/JCO.2011.38.5724
8. Oubari S, Hegenbart U, Schoder R, Steinhardt M, Papathanasiou M, Rassaf T, et al. Daratumumab
in first-line treatment of patients with light chain amyloidosis and Mayo stage IIIb
improves treatment response and overall survival. Haematologica. 2024 Jan;109(1):220–30.
9. Staron A, Zheng L, Doros G, Connors LH, Mendelson LM, Joshi T, et al. Marked progress
in AL amyloidosis survival: a 40-year longitudinal natural history study. Blood Cancer
J. 2021 Aug;11(8):139. doi: https://doi.org/10.1038/s41408-021-00529-w
10. Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez-Naharro A, et
al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018 Aug;39(30):2799–806.
doi: https://doi.org/10.1093/eurheartj/ehx589
11. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a
real-world study using US claims data. Blood Adv. 2018 May;2(10):1046–53. doi: https://doi.org/10.1182/bloodadvances.2018016402
12. Gilstrap LG, Dominici F, Wang Y, El-Sady MS, Singh A, Di Carli MF, et al. Epidemiology
of Cardiac Amyloidosis-Associated Heart Failure Hospitalizations Among Fee-for-Service
Medicare Beneficiaries in the United States. Circ Heart Fail. 2019 Jun;12(6):e005407.
doi: https://doi.org/10.1161/CIRCHEARTFAILURE.118.005407
13. Lauppe R, Liseth Hansen J, Fornwall A, Johansson K, Rozenbaum MH, Strand AM, et al. Prevalence,
characteristics, and mortality of patients with transthyretin amyloid cardiomyopathy
in the Nordic countries. ESC Heart Fail. 2022 Aug;9(4):2528–37. doi: https://doi.org/10.1002/ehf2.13961
14. Castaño A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, et al. Unveiling
transthyretin cardiac amyloidosis and its predictors among elderly patients with severe
aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017 Oct;38(38):2879–87.
doi: https://doi.org/10.1093/eurheartj/ehx350
15. González-López E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M,
Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with
preserved ejection fraction. Eur Heart J. 2015 Oct;36(38):2585–94. doi: https://doi.org/10.1093/eurheartj/ehv338
16. Marchi F, Kessler C, Distefano D, Terzi di Bergamo L, Fumagalli L, Averaimo M, et
al. Prevalence of amyloid in ligamentum flavum of patients with lumbar spinal stenosis.
Amyloid. 2023 Dec;30(4):416–23. doi: https://doi.org/10.1080/13506129.2023.2230516
17. Roberts WC, Waller BF. Cardiac amyloidosis causing cardiac dysfunction: analysis of
54 necropsy patients. Am J Cardiol. 1983 Jul;52(1):137–46. doi: https://doi.org/10.1016/0002-9149(83)90084-X
18. Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance
of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol.
2014 Oct;114(7):1089–93. doi: https://doi.org/10.1016/j.amjcard.2014.07.026
19. Donnellan E, Wazni OM, Hanna M, Elshazly MB, Puri R, Saliba W, et al. Atrial Fibrillation
in Transthyretin Cardiac Amyloidosis: Predictors, Prevalence, and Efficacy of Rhythm
Control Strategies. JACC Clin Electrophysiol. 2020 Sep;6(9):1118–27. doi: https://doi.org/10.1016/j.jacep.2020.04.019
20. Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and
treatment of cardiac amyloidosis: a position statement of the ESC Working Group on
Myocardial and Pericardial Diseases. Eur Heart J. 2021 Apr;42(16):1554–68. doi: https://doi.org/10.1093/eurheartj/ehab072
21. Benz DC, Dorbala S. Multimodality imaging of cardiac amyloidosis. Heart. 2023.
22. Abecasis J, Lopes P, Santos RR, Maltês S, Guerreiro S, Ferreira A, et al. Prevalence
and significance of relative apical sparing in aortic stenosis: insights from an echo
and cardiovascular magnetic resonance study of patients referred for surgical aortic
valve replacement. Eur Heart J Cardiovasc Imaging. 2023 Jul;24(8):1033–42. doi: https://doi.org/10.1093/ehjci/jead032
23. Dorbala S, Cuddy S, Falk RH. How to Image Cardiac Amyloidosis: A Practical Approach.
JACC Cardiovasc Imaging. 2020 Jun;13(6):1368–83. doi: https://doi.org/10.1016/j.jcmg.2019.07.015
24. Mongeon FP, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification
of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovasc
Imaging. 2012 Sep;5(9):897–907. doi: https://doi.org/10.1016/j.jcmg.2012.04.006
25. Poterucha TJ, Elias P, Ruberg FL, DeLuca A, Kinkhabwala M, Johnson LL, et al. False
Positive 99mTc-Pyrophosphate Scanning Leading to Inappropriate Tafamidis Prescriptions.
JACC Cardiovasc Imaging. 2021 Oct;14(10):2042–4. doi: https://doi.org/10.1016/j.jcmg.2021.04.006
26. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy
Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016 Jun;133(24):2404–12.
doi: https://doi.org/10.1161/CIRCULATIONAHA.116.021612
27. Condoluci A, Théaudin M, Schwotzer R, Pazhenkottil AP, Arosio P, Averaimo M, et al. Management
of transthyretin amyloidosis. Swiss Med Wkly. 2021 Oct;151(4142):w30053. doi: https://doi.org/10.4414/SMW.2021.w30053
28. Rauf MU, Hawkins PN, Cappelli F, Perfetto F, Zampieri M, Argiro A, et al. Tc-99m labelled
bone scintigraphy in suspected cardiac amyloidosis. Eur Heart J. 2023 Jun;44(24):2187–98.
doi: https://doi.org/10.1093/eurheartj/ehad139
29. Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and
treatment of cardiac amyloidosis: a position statement of the ESC Working Group on
Myocardial and Pericardial Diseases. Eur Heart J. 2021 Apr;42(16):1554–68. doi: https://doi.org/10.1093/eurheartj/ehab072
30. Roteta Unceta-Barrenechea A, Melero Polo J, Andrés Gracia A, Revilla Martí P, Menao
Guillén S, Lahuerta Pueyo C, et al. Coexistence of Positive 99mTc-DPD Scintigraphy
and Monoclonal Gammopathy: A Frequent Challenge. Zhonghua Minguo Xinzangxue Hui Zazhi.
2022 Mar;38(2):169–74.
31. Phull P, Sanchorawala V, Connors LH, Doros G, Ruberg FL, Berk JL, et al. Monoclonal
gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR).
Amyloid. 2018 Mar;25(1):62–7. doi: https://doi.org/10.1080/13506129.2018.1436048
32. Clerc OF, Cuddy SA, Robertson M, Vijayakumar S, Neri JC, Chemburkar V, et al. Cardiac
Amyloid Quantification Using 124I-Evuzamitide (124I-P5+14) Versus 18F-Florbetapir:
A Pilot PET/CT Study. JACC Cardiovasc Imaging. 2023 Nov;16(11):1419–32. doi: https://doi.org/10.1016/j.jcmg.2023.07.007
33. Benz DC, Gräni C, Antiochos P, Heydari B, Gissler MC, Ge Y, et al. Cardiac magnetic
resonance biomarkers as surrogate endpoints in cardiovascular trials for myocardial
diseases. Eur Heart J. 2023 Dec;44(45):4738–47. doi: https://doi.org/10.1093/eurheartj/ehad510
34. Ioannou A, Massa P, Patel RK, Razvi Y, Porcari A, Rauf MU, et al. Conventional heart
failure therapy in cardiac ATTR amyloidosis. Eur Heart J. 2023 Aug;44(31):2893–907.
doi: https://doi.org/10.1093/eurheartj/ehad347
35. Steinhardt MJ, Cejka V, Chen M, Bäuerlein S, Schäfer J, Adrah A, et al. Safety and
Tolerability of SGLT2 Inhibitors in Cardiac Amyloidosis-A Clinical Feasibility Study.
J Clin Med. 2024 Jan;13(1):283. doi: https://doi.org/10.3390/jcm13010283
36. Dobner S, Bernhard B, Asatryan B, Windecker S, Stortecky S, Pilgrim T, et al. SGLT2
inhibitor therapy for transthyretin amyloid cardiomyopathy: early tolerance and clinical
response to dapagliflozin. ESC Heart Fail. 2023 Feb;10(1):397–404. doi: https://doi.org/10.1002/ehf2.14188
37. Zampieri M, Argirò A, Allinovi M, Perfetto F, Cappelli F. SGLT2i in patients with
transthyretin cardiac amyloidosis, a well-tolerated option for heart failure treatment?
Results from a small, real-world, patients series. Intern Emerg Med. 2022 Jun;17(4):1243–5.
doi: https://doi.org/10.1007/s11739-022-02944-8
38. Dale Z, Chandrashekar P, Al-Rashdan L, Kim M, Masri A, Nazer B. Management Strategies
for Atrial Fibrillation and Flutter in Patients with Transthyretin Cardiac Amyloidosis.
Am J Cardiol. 2021 Oct;157:107–14. doi: https://doi.org/10.1016/j.amjcard.2021.07.028
39. Muchtar E, Gertz MA, Kumar SK, Lin G, Boilson B, Clavell A, et al. Digoxin use in
systemic light-chain (AL) amyloidosis: contra-indicated or cautious use? Amyloid.
2018 Jun;25(2):86–92. doi: https://doi.org/10.1080/13506129.2018.1449744
40. January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr, et al.; American
College of Cardiology/American Heart Association Task Force on Practice Guidelines.
2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation:
a report of the American College of Cardiology/American Heart Association Task Force
on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014 Dec;64(21):e1–76.
doi: https://doi.org/10.1016/j.jacc.2014.03.022
41. Laptseva N, Rossi VA, Sudano I, Schwotzer R, Ruschitzka F, Flammer AJ, et al. Arrhythmic
Manifestations of Cardiac Amyloidosis: Challenges in Risk Stratification and Clinical
Management. J Clin Med. 2023 Mar;12(7):2581. doi: https://doi.org/10.3390/jcm12072581
42. Higgins AY, Annapureddy AR, Wang Y, Minges KE, Lampert R, Rosenfeld LE, et al. Survival
Following Implantable Cardioverter-Defibrillator Implantation in Patients With Amyloid
Cardiomyopathy. J Am Heart Assoc. 2020 Sep;9(18):e016038. doi: https://doi.org/10.1161/JAHA.120.016038
43. Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA, et al.; ESC
Scientific Document Group. 2022 ESC Guidelines for the management of patients with
ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022 Oct;43(40):3997–4126.
doi: https://doi.org/10.1093/eurheartj/ehac262
44. Porcari A, Rossi M, Cappelli F, Canepa M, Musumeci B, Cipriani A, et al. Incidence
and risk factors for pacemaker implantation in light-chain and transthyretin cardiac
amyloidosis. Eur J Heart Fail. 2022 Jul;24(7):1227–36. doi: https://doi.org/10.1002/ejhf.2533
45. Palladini G, Kastritis E, Maurer MS, Zonder J, Minnema MC, Wechalekar AD, et al. Daratumumab
plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results
of ANDROMEDA. Blood. 2020 Jul;136(1):71–80. doi: https://doi.org/10.1182/blood.2019004460
46. Damy T, Garcia-Pavia P, Hanna M, Judge DP, Merlini G, Gundapaneni B, et al. Efficacy
and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical
Trial (ATTR-ACT) and long-term extension study. Eur J Heart Fail. 2021 Feb;23(2):277–85.
doi: https://doi.org/10.1002/ejhf.2027
47. Gillmore JD, Judge DP, Cappelli F, Fontana M, Garcia-Pavia P, Gibbs S, et al.; ATTRibute-CM
Investigators. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy.
N Engl J Med. 2024 Jan;390(2):132–42. doi: https://doi.org/10.1056/NEJMoa2305434
48. Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran,
an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul;379(1):11–21.
doi: https://doi.org/10.1056/NEJMoa1716153
49. Adams D, Tournev IL, Taylor MS, Coelho T, Planté-Bordeneuve V, Berk JL, et al.; HELIOS-A
Collaborators. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated
amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid. 2023 Mar;30(1):1–9.
doi: https://doi.org/10.1080/13506129.2022.2091985
50. Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen
Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul;379(1):22–31.
doi: https://doi.org/10.1056/NEJMoa1716793
51. Coelho T, Marques W Jr, Dasgupta NR, Chao CC, Parman Y, França MC Jr, et al.; NEURO-TTRansform
Investigators. Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy.
JAMA. 2023 Oct;330(15):1448–58. doi: https://doi.org/10.1001/jama.2023.18688
52. Maurer MS, Kale P, Fontana M, Berk JL, Grogan M, Gustafsson F, et al.; APOLLO-B Trial
Investigators. Patisiran Treatment in Patients with Transthyretin Cardiac Amyloidosis.
N Engl J Med. 2023 Oct;389(17):1553–65. doi: https://doi.org/10.1056/NEJMoa2300757
53. Fontana M, Berk JL, Gillmore JD, Witteles RM, Grogan M, Drachman B, et al.; HELIOS-B
Trial Investigators. Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy.
N Engl J Med. 2024 Aug;NEJMoa2409134. doi: https://doi.org/10.1056/NEJMoa2409134
54. Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. CRISPR-Cas9 In
Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med. 2021 Aug;385(6):493–502.
doi: https://doi.org/10.1056/NEJMoa2107454
55. Fontana M, Gilbertson J, Verona G, Riefolo M, Slamova I, Leone O, et al. Antibody-Associated
Reversal of ATTR Amyloidosis-Related Cardiomyopathy. N Engl J Med. 2023 Jun;388(23):2199–201.
doi: https://doi.org/10.1056/NEJMc2304584
56. Garcia-Pavia P, Aus dem Siepen F, Donal E, Lairez O, van der Meer P, Kristen AV, et
al. Phase 1 Trial of Antibody NI006 for Depletion of Cardiac Transthyretin Amyloid.
N Engl J Med. 2023 Jul;389(3):239–50. doi: https://doi.org/10.1056/NEJMoa2303765
57. Schwotzer R, Flammer AJ, Gerull S, Pabst T, Arosio P, Averaimo M, et al. Expert recommendation
from the Swiss Amyloidosis Network (SAN) for systemic AL-amyloidosis. Swiss Med Wkly.
2020 Dec;150(4950):w20364. doi: https://doi.org/10.4414/smw.2020.20364
58. Brouwers S, Heimgartner R, Laptseva N, Aguzzi A, Ehl NF, Fehr T, et al. Historic characteristics
and mortality of patients in the Swiss Amyloidosis Registry. Swiss Med Wkly. 2024 Feb;154(2):3485.
doi: https://doi.org/10.57187/s.3485