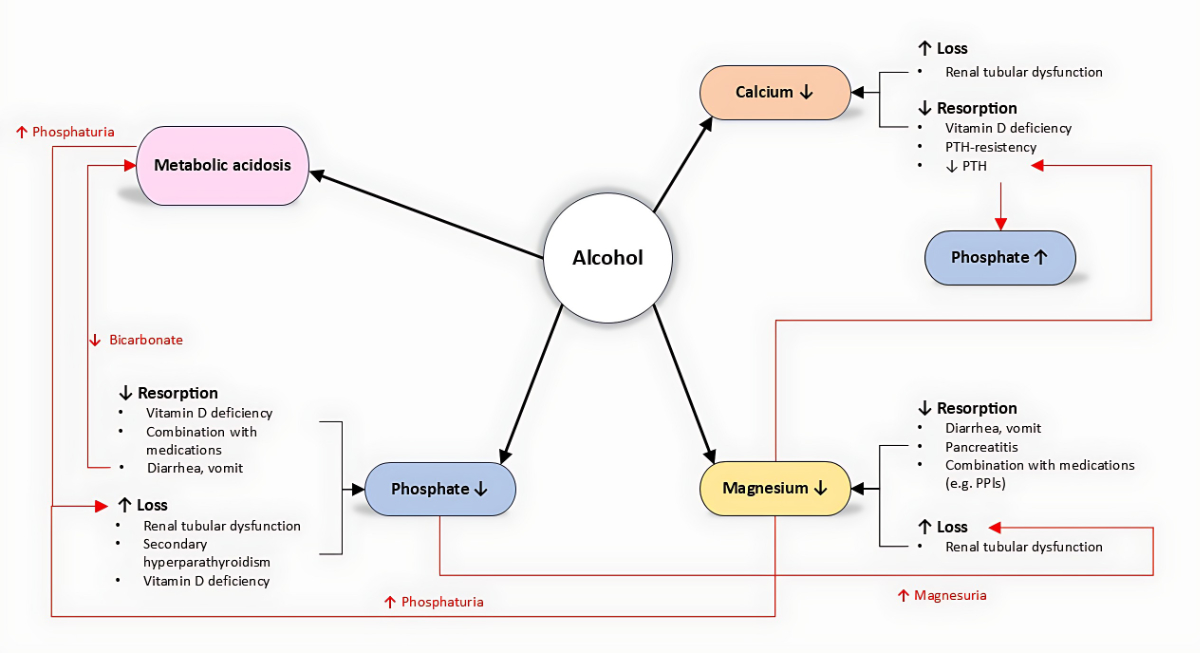
Figure 1Interactions between alcohol, phosphate, calcium, and magnesium.
DOI: https://doi.org/https://doi.org/10.57187/s.4072
Chronic alcohol use disorder is defined as the inability to control alcohol consumption despite negative consequences on health and social conditions. In Switzerland, it is estimated to affect between 220,000 and 330,000 people, the equivalent of 4% of the population [1]. In 2023, nearly 17,000 inpatients were hospitalised in Switzerland because of chronic alcohol use disorder or acute alcohol intoxication [2].
The negative effects on health include neurological damage (Wernicke’s encephalopathy, Korsakoff syndrome, peripheral polyneuropathy, and cognitive decline), increased cardiovascular risk and prevalence of arrhythmias and cardiomyopathy, cancers (especially of the head and neck region and oesophagus), hepatic disorders (ranging from fatty liver disease to liver cirrhosis and hepatocellular carcinoma), and alcoholic pancreatitis. Furthermore, approximately 10% of car accidents in Switzerland (fatal and non-fatal) are associated with acute or chronic alcohol use.
Considering the simultaneously occurring malnutrition and the direct toxic effects of alcohol, electrolyte imbalances are commonly observed in chronic alcohol use disorder. Acute changes in circulating electrolyte levels also occur after alcohol withdrawal, especially in hospitalised patients.
Most commonly, patients with chronic alcohol use disorder develop hyponatraemia. This typically occurs because of hypovolaemia or the beer potomania effect, in which dilutive hyponatraemia develops from excessive fluid intake and low dietary solute intake (sodium, potassium, and protein).
In addition, excessive alcohol intake can cause disturbances in phosphorus, magnesium, and calcium levels. The physiological interactions of these electrolytes and their homeostasis are closely linked (figure 1). An alteration in one can amplify the others, promoting a fatal vicious cycle. This review focuses on the direct and indirect effects of alcohol – as well as alcohol withdrawal – on phosphate, magnesium, and calcium.
Figure 1Interactions between alcohol, phosphate, calcium, and magnesium.
An 82-year-old patient presented to the emergency department after a transient loss of consciousness at home. His medical history included idiopathic lung fibrosis, arterial hypertension, type 2 diabetes mellitus, hypothyroidism, and gastro-oesophageal reflux disease. His medications included candesartan, metoprolol, atorvastatin, insulin degludec, empagliflozin, levothyroxine, and esomeprazole. He reported a daily alcohol intake of approximately 40 g.
Upon arrival, the patient was unconscious (Glasgow Coma Scale: 13 points) and haemodynamically unstable with increased serum lactate levels. The ECG showed atrial fibrillation (heart rate 148/min) with recurrent episodes of non-sustained ventricular tachycardia. Laboratory tests reported hypokalaemia (2.55 mmol/l, normal range: 3.5–4.5 mmol/l), hypocalcaemia (albumin-corrected calcium 1.76 mmol/l, normal range: 2.2–2.6 mmol/l), and hypomagnesaemia (0.27 mmol/l, normal range: 0.66–0.99 mmol/l).
After haemodynamic support, intravenous correction of electrolyte imbalance, and the administration of short-acting beta-blockers, the patient achieved haemodynamic stabilisation and conversion to stable sinus rhythm.
The electrolyte imbalances triggering paroxysmal atrial fibrillation and leading to haemodynamic instability were likely caused by chronic alcohol use and proton pump inhibitors.
Phosphorus is present in the form of phosphate anions (HPO42- or H2PO4-), depending on pH. The normal level in adults lies between 0.80 and 1.30 mmol/l. This may be slightly lower in older adults because of age-related bone loss.
Phosphorus is essential for normal human physiology, with 85% of the body’s phosphorus stored in the bones and teeth. It combines with calcium to form hydroxyapatite, which plays a key role in bone mineralisation. Most intracellular phosphate is bound to organic molecules, forming nucleic acids, adenosine triphosphate, and cofactors for enzymatic reactions (such as nicotinamide adenine dinucleotide phosphate and pyridoxal phosphate). Typical symptoms of acute hypophosphataemia include muscle weakness, respiratory insufficiency, severe cardiac arrhythmias, changes in blood count, and neurological manifestations, such as seizures and coma. Furthermore, hypophosphataemia can worsen the clinical outcomes of alcoholic pancreatitis [3].
Chronic alcohol use disorder leads to hypophosphataemia through both direct and indirect mechanisms. Because of recurrent diarrhoea and vomiting, phosphorus reabsorption is limited and inhibited. The second direct cause of hypophosphataemia is chronic phosphorus excretion via the kidneys, due to alcohol-induced renal tubular dysfunction [4]. In 1993, De Marchi and colleagues showed that patients with chronic alcohol use disorder have a lower excretion threshold, leading to more pronounced phosphorus loss even at lower phosphoraemia levels (1.00 mmol/l compared to 1.19 mmol/l in the general population). Indirectly, alcohol consumption leads to vitamin D deficiency, primarily due to decreased reabsorption and secondarily due to the inhibition of its phosphorylation to the active form, 1,25-dihydroxycholecalciferol. Consequently, secondary hyperparathyroidism can develop and reinforce hypophosphataemia by increased phosphaturia. Metabolic acidosis due to ketogenic metabolism and gastrointestinal bicarbonate loss can lead to increased phosphate excretion in the kidneys as compensatory buffering for the elevated urinary H+ protons [5]. As discussed later, alcohol consumption can lead to hypomagnesaemia, which is suggested to enhance phosphaturia [6]. In 2021, a research group from Japan first described two patients with chronic alcohol use disorder and a constellation of increased fibroblast growth factor 23 (FGF-23) with hypophosphataemia. In both cases, the phosphataemia normalised after a few days of alcohol abstinence, suggesting a reversible, alcohol-induced overproduction of FGF-23 with consequent hyperphosphaturia and hypophosphataemia [7].
Calcium was first isolated in 1808 by Sir Davy and is the fifth most abundant element in the human body, totalling approximately 1000 g. Most of it is complexed as hydroxyapatite and deposited in the bones as a reservoir; only about 1% is found in the intracellular and predominantly the extracellular space. Extracellular calcium is measured as ionised calcium or total calcium, which also includes ions bound to albumin, its main transporter in the blood. The normal ranges vary between 1.05 and 1.30 mmol/l for ionised calcium and 2.2 to 2.5 mmol/l for total calcium. The key factors in calcium homeostasis are parathyroid hormone, vitamin D, and calcitonin. Patients with acute hypercalcaemia typically present with tetany because of increased muscular excitability, paraesthesia, and cramps. Neuropsychiatric symptoms, such as confusion, depression, and delirium, are also frequently observed. The most dangerous manifestations are cardiac repolarisation abnormalities (prolonged QT interval), seizures, and coma.
The most important functions of intracellular calcium are the regulation of nerve impulses, muscular contraction, and hormone secretion. At the extracellular level, calcium plays an important role as a cofactor in the blood coagulation cascade and in maintaining the transmembrane potential.
Hypocalcaemia is typically observed in patients with chronic alcohol use disorder. This can arise through various mechanisms causing either reduced absorption or increased loss. As mentioned above, patients with chronic alcohol use disorder often develop vitamin D deficiency, which can lead to hypocalcaemia by impairing intestinal reabsorption. Moreover, alcohol-mediated tubular dysfunction increases calciuria [4]. Calciuria can also be directly increased by a transient hypoparathyroid state caused by acute alcohol consumption, as described in 1991 [8]. This condition is further aggravated by hypomagnesaemia, as magnesium is required for PTH secretion [9]. Acute pancreatitis, a frequent complication of chronic alcohol use disorder, can lead to hypocalcaemia with hypoparathyroidism, which also has prognostic value in pancreatitis [10]. The exact cause of hypocalcaemia in acute pancreatitis remains unclear, but it has been hypothesised that calcium forms a complex with peripancreatic fatty acids, which are digested from the pancreatic enzymes [11]. Hypomagnesaemia also impairs the secretion of, and increases resistance to, parathyroid hormone (PTH), leading to a picture of the abovementioned transient hypoparathyroidism [12].
Sir Davy also isolated magnesium in 1808. Most of the approximately 25 g of magnesium in the body is stored in the bones and muscles, with the majority located in the mineral matrix of bone. Normal circulating magnesium levels are between 0.7 and 1.0 mmol/l.
Physiologically, magnesium is a cofactor in various enzymatic reactions, such as ATP and nucleic acid synthesis, and intracellular signal transduction. Moreover, magnesium directly affects PTH and vitamin D synthesis, playing a role in the homeostasis of calcium and phosphorus. At the cellular level, it plays a role in immune system regulation, and lower magnesium levels can stimulate the inflammatory cascade. Neuromuscular hyperexcitability, a typical clinical manifestation of acute magnesium deficiency, presents with spasms, seizures, tremor, or nystagmus. Cardiovascular complications include arrhythmia, hypertension, and increased risk of atherosclerosis.
Hypomagnesaemia is a classic condition found in patients with chronic alcohol use disorder and is partially explained by reduced magnesium intake. As seen with phosphorous and calcium, other mechanisms lead to either decreased absorption or increased loss. Decreased absorption can occur following vomiting and diarrhoea. Acute pancreatitis can also cause hypomagnesaemia in a similar way to that discussed for hypocalcaemia [10]. Chronic pancreatitis with steatorrhoea can lead to the saponification of magnesium in the gastrointestinal tract, which prevents the body from absorbing it. Proton pump inhibitors, typically prescribed to patients with chronic alcohol use disorder and concomitant gastritis, can also decrease magnesium absorption. Hypermagnesuria in patients with chronic alcohol use disorder may also result from renal tubular dysfunction due to ethanol and hypophosphataemia [13, 14]. The resulting hypomagnesaemia further aggravates renal phosphate loss, creating a vicious cycle and explaining the frequent coexistence of these disorders.
Alcohol withdrawal can worsen hypophosphataemia, resulting in severe, life-threatening imbalances. The main pathogenic mechanism is an intracellular shift of phosphate anions, partly due to the increased production of stress-related hormones, such as adrenaline, during withdrawal. In addition, concomitant respiratory alkalosis following hyperventilation in alcohol withdrawal raises pH, which accentuates intracellular phosphate influx. Moreover, infusions with glucose and the consequent production of insulin can stimulate phosphorus uptake into cells as a catalyst of glycolysis. Because of this effect, glucose-containing fluids should be avoided in patients with hypophosphataemia who have chronic alcohol use disorder at admission, unless otherwise indicated (e.g. in those with hypoglycaemia). Similar mechanisms might be involved in hypomagnesaemia. Finally, the threshold level for phosphate excretion increases after four weeks of abstinence [4].
Little is known about calcium levels during alcohol withdrawal. Hypothetically, the stress response and increased release of catecholamines may stimulate PTH secretion. However, the exact effects of adrenergic overactivation on calcium homeostasis remain unknown. A recent study on animals showed that adrenaline caused an increase in circulating calcium levels [15], although clinical data are conflicting. Several studies have shown a reduction in ionised calcium after exercise, whereas others have reported no change or even an increase in calcium levels [16]. The exact response of the mineral to increased stress hormone secretion is unknown.
Regarding the measurement of calcium levels, it is important to note that, as patients with chronic alcohol use disorder are often malnourished, hypalbuminaemia may be present. Hypalbuminaemia results in pseudohypocalcaemia without clinical relevance. To address the physiological impact of clinically significant hypocalcaemia, ionised calcium should be measured [17].
The normalisation of the pH via correction of metabolic acidosis can cause an intracellular shift of magnesium, potentially worsening hypomagnesaemia or unmasking latent hypomagnesaemia [18]. Glucose infusion as fluid therapy in patients with chronic alcohol use disorder and increased release of stress hormones can also worsen hypomagnesaemia by shifting magnesium ions into the cells.
Hypokalaemia, hypomagnesaemia, and hypocalcaemia, the main laboratory findings in the clinical vignette, are all associated with increased mortality [19]. Hypophosphataemia is also well-described as a predictor of higher mortality, especially in critically ill patients [20]. Hypophosphataemia is found in up to 50% of patients with chronic alcohol use disorder, and hypomagnesaemia is observed in up to 27% [18, 21]. Considering the relatively high prevalence of chronic alcohol use disorder, we believe that the pathophysiological mechanisms and interconnections of this topic are highly relevant in daily clinical practice.
The first important aspect is that the risk of developing electrolyte imbalance in patients with chronic alcohol use disorder increases in a dose-dependent manner [14, 18]. However, exact data to determine how much the prevalence increases with increased alcohol consumption are lacking. Both the amount and duration of alcohol intake influence the severity of the clinical and laboratory features. A patient with chronic alcohol use disorder may develop a chronic steady state in a pathological laboratory range without any clinical signs of an electrolyte imbalance. By contrast, acute alcohol intoxication with an acute electrolyte imbalance may lead to severe symptoms or clinical findings, ranging from seizures to arrhythmias.
Secondly, as described above, all electrolyte imbalances can be directly caused by alcohol. In other words, even young patients with a normal nutritional status and no comorbidities may have electrolyte imbalances. Comorbidities and medications might further promote these imbalances.
The risk of hypophosphataemia in patients with diabetic ketoacidosis is well known, as the administration of insulin stimulates the shift of phosphate into the intracellular space. Few studies have investigated the correlation between phosphate levels and insulin resistance. In a retrospective cohort of 177 patients with diabetes, Carnevale et al. showed that those with insulin resistance had slightly increased phosphate levels [22]. However, it is unclear whether higher phosphate levels are a cause or consequence of insulin resistance – both processes likely reinforce each other as part of a vicious cycle. It is also important to note that older patients with chronic alcohol use disorder tend to be malnourished, which can lead to increased insulin sensitivity. This may stimulate the intracellular translocation of phosphate and thus promote hypophosphataemia. To our knowledge, no data are available on phosphate levels in patients with type 2 diabetes mellitus and increasing insulin sensitivity.
Moreover, insulin stimulates renal reabsorption of magnesium, especially in the distal convoluted tubule, suggesting that hypomagnesaemia may develop in a state of insulin deficiency [23]. Relevant derangements of magnesium homeostasis require a poorly controlled diabetes mellitus. One study found that the blood calcium levels were not significantly different between patients with insulin resistance and insulin-sensitive individuals [22]. Hypocalcaemia and hypercalcaemia are rarely seen as a complication of well-controlled diabetes mellitus or as a side effect of insulin therapy, the main exception being hypercalcaemia following dehydration due to hyperglycaemia and concomitant acute renal failure [24]. In our case, we believe that neither the well-controlled type 2 diabetes mellitus nor the insulin therapy played a major role in the development of the electrolyte imbalances.
SGLT2 inhibitors, effective reno- and cardioprotective agents, are increasingly used in daily practice. Because sodium is less easily reabsorbed in the proximal tubule after the administration of an SGLT2 inhibitor, its higher levels in the urine stimulate sodium-phosphate co-transporter (NaPi2a), increasing the reabsorption of phosphate and explaining the slightly higher phosphataemia [25]. It is also well described that SGLT2 inhibitors can slightly increase serum magnesium in patients with type 2 diabetes mellitus; for instance, 10 mg of empagliflozin led to a mean increase of 0.04 mmol/l compared with placebo in a large meta-analysis [26]. It is thus very unlikely that empagliflozin was important in the development of the electrolyte imbalances in the case vignette.
Thyroid hormones also affect electrolyte metabolism, the most prominent examples being hypercalcaemia in hyperthyroidism (high bone turnover) and hyponatraemia in hypothyroidism (increased water retention). However, the patient’s substitution therapy with levothyroxine was adequate, so no electrolyte imbalance was expected.
In Denmark, one-fifth of patients with chronic alcohol use disorder receive regular medication with a proton pump inhibitor [27]. A prospective cohort study with almost 10,000 patients showed a significant association between the use of proton pump inhibitors and the development of hypomagnesaemia, mainly because a higher pH in the gastrointestinal lumen inhibits the reabsorption of magnesium [28, 29]. In patients receiving the common combination of proton pump inhibitors and diuretics, the risk of hypomagnesaemia is even higher [30].
The presentation of the patient in the vignette describes a typical constellation in chronic alcohol use disorder. Electrolyte imbalances – namely hyponatraemia, hypokalaemia, hypophosphataemia, hypomagnesaemia, and hypocalcaemia – are common in patients with chronic alcohol use disorder and alcohol withdrawal syndrome, particularly in older, malnourished patients with comorbidities and multiple medications. These conditions require prompt recognition, laboratory monitoring, and replacement therapy. We suggest measuring phosphate, magnesium, and ionised calcium in patients with chronic alcohol use disorder and those experiencing alcohol withdrawal, especially if they present with high-risk constellations as described above.
In addition to the common hyponatraemia, excessive and chronic alcohol consumption can lead to disturbances in phosphorus, magnesium, and calcium homeostasis.
Due to the close physiological interactions between phosphorus, magnesium, and calcium, alterations of one or more of them can result in a life-threatening vicious cycle.
Electrolyte imbalances are common in alcohol withdrawal and require careful monitoring and management.
This study received no funding.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. No potential conflict of interest related to the content of this manuscript was disclosed.
1. Kuendig H. Estimation du nombre de personnes alcoolo-dépendantes dans la population helvétique (Rapport de recherce No 56). Lausanne: Addiction Info Suisse; 2010., Available from https://www.suchtmonitoring.ch/docs/library/kuendig_herve_ii2ajqwt12iq.pdf
2. Schweizerische Eidgenossenschaft - Bundesamt für Statistik. Medizinische Statistik der Krankenhäuser: Anzahl Fälle und durchschnittliche Aufenthaltsdauer (DAD) nach Altersklasse und Diagnosekode. Dezember 2024. Available from: https://www.bfs.admin.ch/bfs/de/home/statistiken/kataloge-datenbanken.assetdetail.33787452.html
3. Wagner J, Hernandez-Blanco Y, Yu A, Garcia-Rodriguez V, Mohajir W, Goodman C, et al. Hypophosphatemia Is More Common and Is Prognostic of Poorer Outcomes in Severe Alcoholic Pancreatitis. Pancreas. 2021 Nov-Dec;50(10):1440–4. doi: https://doi.org/10.1097/MPA.0000000000001952
4. De Marchi S, Cecchin E, Basile A, Bertotti A, Nardini R, Bartoli E. Renal tubular dysfunction in chronic alcohol abuse—effects of abstinence. N Engl J Med. 1993 Dec;329(26):1927–34.
5. Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol. 2014 Sep;9(9):1627–38.
6. Ginn HE, Shanbour LL. Phosphaturia in magnesium-deficient rats. Am J Physiol. 1967 Jun;212(6):1347–50.
7. Hidaka N, Kato H, Koga M, Katsura M, Oyama Y, Kinoshita Y, et al. Induction of FGF23-related hypophosphatemic osteomalacia by alcohol consumption. Bone Rep. 2021 Oct;15:101144.
8. Laitinen K, Lamberg-Allardt C, Tunninen R, Karonen SL, Tähtelä R, Ylikahri R, et al. Transient hypoparathyroidism during acute alcohol intoxication. N Engl J Med. 1991 Mar;324(11):721–7.
9. Na D, Tao G, Shu-Ying L, Qin-Yi W, Xiao-Li Q, Yong-Fang L, et al. Association between hypomagnesemia and severity of primary hyperparathyroidism: a retrospective study. BMC Endocr Disord. 2021 Aug;21(1):170.
10. Robertson GM Jr, Moore EW, Switz DM, Sizemore GW, Estep HL. Inadequate parathyroid response in acute pancreatitis. N Engl J Med. 1976 Mar;294(10):512–6.
11. Ryzen E, Rude RK. Low intracellular magnesium in patients with acute pancreatitis and hypocalcemia. West J Med. 1990 Feb;152(2):145–8.
12. Rude RK, Oldham SB, Singer FR. Functional hypoparathyroidism and parathyroid hormone end-organ resistance in human magnesium deficiency. Clin Endocrinol (Oxf). 1976 May;5(3):209–24.
13. Laitinen K, Tähtelä R, Välimäki M. The dose-dependency of alcohol-induced hypoparathyroidism, hypercalciuria, and hypermagnesuria. Bone Miner. 1992 Oct;19(1):75–83.
14. Baj J, Flieger W, Teresiński G, Buszewicz G, Sitarz R, Forma A, et al. Magnesium, Calcium, Potassium, Sodium, Phosphorus, Selenium, Zinc, and Chromium Levels in Alcohol Use Disorder: A Review. J Clin Med. 2020 Jun;9(6):1901. doi: https://doi.org/10.3390/jcm9061901
15. Shkurashivska S, Ersteniuk H. The Effect of Adrenaline on the Mineral and Trace Element Status in Rats. Open Life Sci. 2019 May;14(1):158–64.
16. Lombardi G, Ziemann E, Banfi G, Corbetta S. Physical Activity-Dependent Regulation of Parathyroid Hormone and Calcium-Phosphorous Metabolism. Int J Mol Sci. 2020 Jul;21(15):5388.
17. Kenny CM, Murphy CE, Boyce DS, Ashley DM, Jahanmir J. Things We Do for No Reason™: Calculating a “Corrected Calcium” Level. J Hosp Med. 2021 Aug;16(8):499–501.
18. Palmer BF, Clegg DJ. Electrolyte Disturbances in Patients with Chronic Alcohol-Use Disorder. N Engl J Med. 2017 Oct;377(14):1368–77.
19. Ayhan YE, İlerler EE, Sosyal D, Bektay MY, Karakurt S, Daşkaya H, et al. Assessment of drug-induced electrolyte disorders in intensive care units: a multicenter observational study. Front Med (Lausanne). 2024 Jun;11:1343483.
20. Geerse DA, Bindels AJ, Kuiper MA, Roos AN, Spronk PE, Schultz MJ. Treatment of hypophosphatemia in the intensive care unit: a review. Crit Care. 2010;14(4):R147.
21. Vanoni FO, Milani GP, Agostoni C, Treglia G, Faré PB, Camozzi P, et al. Magnesium Metabolism in Chronic Alcohol-Use Disorder: Meta-Analysis and Systematic Review. Nutrients. 2021 Jun;13(6):1959.
22. Carnevale V, Nieddu L, Scillitani A, Tinti MG, Eller-Vainicher C, Cosso R, et al. Calcium-phosphate homeostasis and insulin resistance in men. Nutr Metab Cardiovasc Dis. 2024 Feb;34(2):353–9.
23. Gommers LM, Hoenderop JG, Bindels RJ, de Baaij JH. Hypomagnesemia in Type 2 Diabetes: A Vicious Circle? Diabetes. 2016 Jan;65(1):3–13.
24. Liamis G, Liberopoulos E, Barkas F, Elisaf M. Diabetes mellitus and electrolyte disorders. World J Clin Cases. 2014 Oct;2(10):488–96.
25. Palmer BF, Clegg DJ. Kidney-Protective Effects of SGLT2 Inhibitors. Clin J Am Soc Nephrol. 2023 Feb;18(2):279–89.
26. Tang H, Zhang X, Zhang J, Li Y, Del Gobbo LC, Zhai S, et al. Elevated serum magnesium associated with SGLT2 inhibitor use in type 2 diabetes patients: a meta-analysis of randomised controlled trials. Diabetologia. 2016 Dec;59(12):2546–51.
27. Llorente C, Jepsen P, Inamine T, Wang L, Bluemel S, Wang HJ, et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat Commun. 2017 Oct;8(1):837.
28. Kieboom BC, Kiefte-de Jong JC, Eijgelsheim M, Franco OH, Kuipers EJ, Hofman A, et al. Proton pump inhibitors and hypomagnesemia in the general population: a population-based cohort study. Am J Kidney Dis. 2015 Nov;66(5):775–82.
29. Adomako EA, Yu AS. Magnesium Disorders: core Curriculum 2024. Am J Kidney Dis. 2024 Jun;83(6):803–15.
30. Rosner MH, Ha N, Palmer BF, Perazella MA. Acquired Disorders of Hypomagnesemia. Mayo Clin Proc. 2023 Apr;98(4):581–96.