Blastic plasmacytoid dendritic cell neoplasm: a Swiss case series of a very rare disease
and a structured review of the literature
DOI: https://doi.org/https://doi.org/10.57187/s.3885
Ramona Meier-Lienhardab,
Cosima Suterc,
Thomas Pabstd,
Felicitas Hitze,
Jakob R. Passwegf,
Olivier Spertinig,
Nathan Cantonih,
Daniel Betticheri,
Lucas Simeonj,
Michael Medingerk,
Stefanie Hayozl,
Adrian Schmidtb
a Department of Internal Medicine,
Clinic for Medical Oncology, Lucerne Cantonal Hospital, Lucerne, Switzerland
b Department of Internal Medicine,
Clinic for Medical Oncology and Hematology, Municipal Hospital Zurich Triemli, Zurich,
Switzerland
c Clinic for Medical Oncology and
Hematology, Zurich University Hospital, Zurich, Switzerland
d Department of Medical Oncology,
Inselspital, Bern University Hospital, Bern, Switzerland
e Division of Hematology, Kantonsspital St Gallen, St Gallen, Switzerland
f Hematology Division, Basel University
Hospital, Basel, Switzerland
g Service of Hematology, Department of
Oncology, Lausanne University Hospital, Lausanne, Switzerland
h Division of Hematology, Kantonsspital
Aarau AG, Aarau, Switzerland
i Division of Oncology and Hematology, HFR
Fribourg, Fribourg, Switzerland
j Department of Internal Medicine,
Clinic for Hematology, Lucerne Cantonal Hospital, Lucerne, Switzerland
k Department of Internal Medicine,
Clinic for Medical Oncology, Hematology and Palliative Care, Diakonie-Klinikum
Schwäbisch Hall GmbH, Schwäbisch Hall, Germany and University of Basel, Basel,
Switzerland
l SAKK
Competence Center, Bern, Switzerland
Summary
INTRODUCTION:
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a very rare disease, with
unique diagnostic challenges and often dismal outcome. There are no widely
accepted treatment guidelines available. Lymphoma-like regimens with or without
autologous or allogenic transplantation were the cornerstone of most therapeutic
concepts. A few years ago, the CD123-directed immunoconjugate tagraxofusp emerged
as a new valuable treatment option. The goal of our research was to collect
available data on BPDCN-patients treated at large centres in Switzerland and
worldwide and to draw conclusions regarding the incidence, clinical
presentation, prognostic factors and therapeutic strategies.
METHODS: We
collected data from BPDCN patients from leading Swiss haemato-oncology centres
from 2005 to 2022. Furthermore, we reviewed and analysed the published
literature (cohorts and case reports in peer-reviewed journals) from 1997 to
2020 (structured review of the literature).
RESULTS: We
identified 115 international publications including 600 patients from all over
the world. Most of them had very small sample sizes (only ten papers with more
than ten patients) and all but one were retrospective or observational respectively.
Most included patients were Europeans
(n = 385, 64%) and Asians (n = 120, 20%), followed by Americans (n = 90, 15%)
and patients from Australia/New Zealand (n = 3) and Africa (n = 2). BPDCN was
more common in men with a predominance of 3:1. The median age (n = 414) at
diagnosis was 66.5 years ranging from one month to 103 years. Newly diagnosed women
were significantly younger than men (median: 62 vs 67 years, mean: 53.4 vs
59.3 years, p = 0.027) and less often had bone marrow infiltration and affected lymph
nodes. Upfront allogenic transplantation as well as ALL regimens performed best,
with response to first-line therapy clearly associated with better overall
survival. The Swiss cohort contained 26 patients (23 males and 3 females) over 18
years (2005–2022). The median age at diagnosis was 68.5 years (range: 20–83). Ten
patients underwent upfront stem cell transplantation (seven allogenic and three
autologous), at least trending towards a better overall survival than other
therapies. With a follow-up of 8 years, the median overall survival was 1.2
years. Eight patients in this cohort were treated with tagraxofusp, which became
available in 2020 and was approved by Swissmedic in 2023.
CONCLUSIONS: Our study confirms that BPDCN is a very
rare and difficult-to-treat disease. Underdiagnosis and underreporting in the
literature pose further challenges. Symptoms at presentation seem to differ slightly
between sexes and reaching a complete remission after first-line treatment remains
crucial for a prolonged overall survival. Effective treatment protocols in
first line include transplantation regimens (mainly allogenic, potentially also
autologous) as well as ALL protocols. In order to understand the significance
of tagraxofusp as a bridge to transplant or as a continuous monotherapy in elderly
patients, further evaluation with longer follow-up periods is required. In
general, analysis of the Swiss patients confirmed the results from the
worldwide cohort.
Introduction
Blastic
plasmacytoid dendritic cell neoplasm (BPDCN) is a very rare disease with an annual
incidence of about 1 case / 2.5 million [1]. Patients usually present in their seventh
decade; however all age groups are represented. A slight male predominance is
well known with no ethnic predilection [2].
Uncertainties
about the histogenesis of BPDCN have led to several changes in nomenclature in
the past 25 years. At first, a T-cell origin was proposed based on the CD4
positivity of the cell. Hence BPDCN was named “plasmacytoid T-cell lymphoma”. When
the expression of CD56 and a lack of other T-cell markers on the cells were later
documented, natural killer cells were regarded as the cells of origin. Accordingly,
the WHO classification of 1999 classified the CD4+/CD56+ neoplasm as blastic natural
killer cell lymphoma [3]. A few years later, Chaperot et al. [4] were able to demonstrate
that these neoplastic cells expressing CD4/CD56 in the absence of typical
markers of B-cell and T-cell lineage were derived from precursors of plasmacytoid
dendritic cells. Based on these findings the term BPDCN was established and it
has been used since 2008. The WHO classification of 2008 categorised BPDCN as
an entity of “acute myeloid leukaemia and related precursor neoplasms”. In the revised
classification of 2016, BPDCN was finally classified as a separate entity [5]. The
5th and most recent edition of the WHO classification (2022) brought new changes,
specifically revised diagnostic criteria. Currently, the diagnosis of BPDCN is
established when one of the following two sets of criteria is met:
- expression of
CD123 and one other pDC (plasmacytoid dendritic) marker (TCF4, TCL1,
CD303, CD304) in addition
to CD4 and/or CD56, or
- expression of any three pDC
markers and absence of expression of all expected negative markers (CD3,
CD14, CD19, CD34, lysozyme, myeloperoxidase) [6].
The clinical presentation is heterogeneous, but skin involvement at
presentation is common, varying from nodular lesions to patch-plaques or even bruise-like
patterns. In advanced stages of the disease, lymph node involvement, bone
marrow infiltration and leukaemic transformations with marked cytopenia occur
regularly [2].
The diagnosis
is difficult and often missed or at least delayed. The cornerstone of the diagnosis
is immunohistochemical work-up of skin biopsy or other affected tissues. BPDCN
may be considered in cases of a morphologically diffuse, monomorphous infiltration
of dermis with extension to subcutaneous fat (but not the epidermis however) of
medium-sized blast cells with irregular nuclei and fine chromatin, some
nucleoli, scant and agranular cytoplasm, variable mitosis, and the absence of
angioinvasion and coagulative necrosis [5]. The diagnosis is confirmed by the expression
of the typical markers, as mentioned above.
Nevertheless,
differentiating neoplastic from reactive plasmacytoid dendritic cells is often very
challenging, even for experienced haematologists. Several molecular and
cytogenetic abnormalities have been reported, but none of them are unique to
BPDCN. The diagnostic work-up/staging is completed by bone marrow examination and
a PET/CT (currently not mandatory) or CT scan (with particular emphasis on
lymph nodes and extramedullary disease [7]. Given the high incidence of occult
cerebrospinal fluid involvement, a spinal fluid examination (including cytology
and flow cytometry) is recommended in all patients [8].
Treatment
of BPDCN is difficult and demanding, even though
several treatment recommendations and position papers are now available [7–9].
In the
past, most patients were treated with different chemotherapeutic regimens
(acute lymphoblastic leukaemia-like, acute myeloid leukaemia-like,
lymphoma-like). Despite high initial response rates, most patients relapse
early and have dismal outcomes as reflected by the low median survival of only
slightly above 12 months. This clinical course is usually independent of the
initial pattern of the disease. Expression of CD303 and elevated Ki-67 might be
associated with a longer survival [9]. Patients with a leukaemic stage have a worse
prognosis with a median survival of 8.7 months [11]. Unequivocal curative
concepts do not exist, even if long-term remissions in adults have been reported
in selected cases with autologous or allogeneic stem cell transplantation [12].
Given that many patients are elderly, have additional comorbidities and present
with advanced disease, therapeutic options are limited, especially regarding
intensive approaches such as transplantation.
A promising
new agent became available in 2019: tagraxofusp (Elzonris®, Stemline Therapeutics B.V.), a recombinant fusion protein consisting
of human interleukin-3 fused to truncated diphtheria toxin that targets cells
expressing CD123, leading to apoptosis [13]. Administration is by the intravenous
route on days 1–5 of any 21-day cycle. Tagraxofusp is approved by the FDA, the EMA
and Swissmedic and reimbursed by healthcare providers in Switzerland.
The goals
of our study were to gain an overview of the prevalence, treatment and outcomes
of Swiss patients with BPDCN in the last 18 years; to collect the available
literature for the diagnosis and treatment of BPDCN; and to draw – in a
cautious manner – conclusions about optimal management of this disease.
Materials and methods
We
conducted extensive research of the available literature, mainly using the databases
PubMed and Google Scholar as well as the relevant references in UpToDate. Our
search terms were “BPDCN” and “blastic plasmacytoid dendritic cell neoplasm”. Papers
in English, German, French and Italian were considered. For every paper, we also
checked the references to obtain further information about cohort studies or
case reports involving patients with BPDCN. We obtained data published between 1
January 1997 and 31 December 2020. The key features of papers were entered in Excel
spreadsheets, especially the following parameters: first author, type and
geographic region of study, number of patients, age and sex distributions,
clinical presentation at diagnosis, therapeutic regimen used, use of novel or
experimental drugs, response rates, evolution and survival. Therapeutic
regimens were divided into eight categories as follows: allogeneic
transplantation (ALLO), autologous transplantation (AUTO), ALL-like without
transplantation (ALL), AML-like without transplantation (AML), AL-like without
further information without transplantation (AL NOS), lymphoma-like (LL),
others (O), unknown (U). Table 1 lists the therapeutic regimens included in
each category, under consideration of sometimes blurred borders.
Table 1Categories of therapeutic regimens.
| Allogeneic
stem cell transplantation (ALLO) |
Induction therapies containing cytarabine/anthracycline ±
intrathecal cytarabine, CHOP, MTX, etoposide, cytarabine |
| Autologous stem cell transplantation (AUTO) |
Induction therapy with CHOP |
| Acute lymphatic leukaemia (ALL) |
Ifosfamid, etoposide, prednisone ± cytarabine ± MTX ±
L-asparaginase ± mitoxantrone |
| Hyper-CVAD |
| VPDL (vincristine, methylprednisolone, daunorubicin,
L-asparaginase) ± MTX |
| LALA 94 protocol |
| GRAALL 2003 protocol |
| Acute myeloid leukaemia (AML) |
Cytarabine/anthracycline ± intrathecal MTX ± etoposide ±
mitoxantrone ± fludarabine ± lomustine ± vincristine |
| LAM-90 protocol |
| Acute
leukaemia not otherwise specified (AL NOS) |
No specified protocols |
| Lymphoma-like (LL) |
CHOP-like (COP ± anthracycline ± rituximab ± etoposide ± bleomycin ± chlorambucil
± MTX ± mitoxantrone ± intrathecal MTX) |
| Hyper-CVAD |
| ABVD |
| IE, ICE or IME (MTX instead of carboplatin) |
| ESHAP |
| Other therapeutic options (O) in
alphabetical order |
Azacytidine |
| Bendamustine |
| Best supportive care / no
treatment |
| Cytarabine ± mitoxantrone ± etoposide |
| Daratumumab |
| EC
(epirubicin, cyclophosphamide) |
| Hydroxyurea |
| Interferon alpha ±
bexarotene |
| POMP-like |
| Radiotherapy ±
etoposide |
| Steroid
monotherapy |
| Tagraxofusp |
| Unspecified
chemotherapy |
| VAD
(vincristine, anthracycline, dexamethasone) plus radiotherapy |
| Venetoclax |
| VRd
(bortezomib, lenalidomide, dexamethasone) |
As we did
not request the original raw data, our findings are based solely on the
published information. The gathered information was analysed using descriptive
as well as non-parametric statistics, the latter especially to compare outcomes
of different therapeutic approaches. We quickly realised that investigating our
pre-defined parameters was more difficult than expected, due to missing data and the inclusion of several patients in more than
one report. In contrast to the descriptive analysis, exploratory statistics were
only done in patient cases with availability of complete information as needed,
maybe allowing a certain bias. The number of included patients is declared in a
transparent manner on every separate analysis.
Statistical
analyses were performed with Excel (Microsoft, Redmond, WA, USA), R 4.2.1 (www.r-project.org) and Vassarstats (Poughkeepsie, NY,
USA). Categorical variables were compared between groups using Fisher’s
exact test. Continuous variables were compared between groups using Student’s t-test
for the international cohort and the Wilcoxon rank-sum test for the Swiss
cohort. In the Swiss cohort, overall survival was calculated from diagnosis to
death. Patients with no recorded death were censored at the date they were last
known to be alive. In the structured review of the literature, we took the
survival data from the original publications as far as possible. Overall
survival was illustrated using the Kaplan-Meier methods and compared between
groups using log-rank tests. The influence of covariables on overall survival
was analysed using uni- and multivariate Cox regression models. For multivariate
models, clinically relevant covariables were included as well as the therapy
with the lowest hazard ratio, as due to collinearity only one therapy could be
included.
In order to
obtain an adequate overview of the Swiss BPDCN landscape, we contacted the
leading local haemato-oncology centres asking for their data from BPDCN
patients since 2005. The following centres (in alphabetical order) provided
information: Cantonal Hospital Aarau, University Hospital Basel, Instituto
Oncologico della Svizzera Italiana Bellinzona, University Hospital Inselspital
Bern, Cantonal Hospital Chur, Cantonal Hospital Fribourg, University Hospital
Geneva (HUG), University Hospital Lausanne (CHUV), Cantonal Hospital Baselland
Liestal, Cantonal Hospital Lucerne, Cantonal Hospital St Gallen, Hirslanden
Zurich, Municipal Hospital Zurich, University Hospital Zurich. For each patient,
we collected data on the initial clinical presentation, histopathological
features, the therapeutic regimen selected at first and in case of relapse,
remission status and overall survival as well as epidemiological data (sex, age
at diagnosis). Of note, the same eight therapeutic categories were maintained
as in the international structured review of the literature. Transplant (ALLO
or AUTO) as first-line treatment means: The decision to do an allogenic
(autologous) stem cell transplantation was taken before starting treatment with
induction therapy (and not at the time point when 1st line therapy failed, then
ALLO or AUTO were counted as 2nd line treatment). All data were provided in an
anonymised form and the study was approved by the local ethics committee of
Nordwest- und Zentralschweiz EKNZ (Nr. 2016/271). A study protocol was not
prepared. All study data are available to those with a legitimate interest
(Open Science).
Results
Swiss cohort
We obtained
data from 26 patients diagnosed with blastic plasmacytoid dendritic cell
neoplasm (BPDCN) in the Swiss centres since 2005. Another two patients (both
female) had to be excluded from analysis due to missing/denied general consent
(table 2).
Table 2Overview of the Swiss cohort with 26 patients.
| Patient # |
Sex / age (years) |
Clinical picture at diagnosis |
Therapeutic regimen after diagnosis |
First-line
therapy → Response (→ Relapse) |
Overall survival (months) |
| Skin |
Lymph node |
Bone marrow |
Peripheral blood |
| 1 |
M/53 |
yes |
no |
yes |
no |
ALLO-SCT (AML protocol) |
ALLO → CR |
100.9* |
| 2 |
M/61 |
yes |
no |
no |
no |
ALLO-SCT (AML protocol) |
ALLO → CR |
75.5** |
| 3 |
M/20 |
yes |
no |
no |
no |
CODOX-M IVAC
(LL protocol) |
LL → CR →
Relapse |
6.2 |
| 4 |
M/78 |
yes |
yes |
yes |
yes |
Pralatrexate (LL) |
LL → PD |
8.1 |
| 5 |
M/40 |
no |
no |
yes |
no |
ALLO-SCT (cytarabine, idarubicin) |
ALLO → CR → Relapse |
30 |
| 6 |
F/75 |
no |
no |
yes |
yes |
Azacytidine (AML protocol) |
AML → PD |
7.1 |
| 7 |
M/68 |
no |
no |
yes |
no |
GMALL
elderly (ALL protocol) |
ALL → PD |
3.9 |
| 8 |
M/74 |
yes |
no |
no |
no |
CHOEP (LL) |
LL → PR → PD |
10.8 |
| 9 |
M/44 |
no |
yes |
no |
no |
ALLO-SCT (hyper-CVAD, HD-MTX/cytarabine) |
ALLO → CR |
97.8* |
| 10 |
M/81 |
yes |
yes |
yes |
yes |
3 cycles tagraxofusp |
O*** → CR → Relapse |
10.3** |
| 11 |
M/72 |
yes |
yes |
yes |
yes |
AUTO-SCT (GRAALL-2014) |
AUTO → CR |
45.6* |
| 12 |
M/47 |
yes |
yes |
yes |
yes |
1 cycle hd cytarabine, 2 cycles
tagraxofusp (complete remission after 1 cycle) |
O*** → CR → Relapse |
1.9 |
| 13 |
F/69 |
yes |
yes |
yes |
no |
2 cycles cytarabine/daunorubicin (AML protocol) |
AML → PD |
4.6 |
| 14 |
M/71 |
yes |
no |
no |
no |
None (only skin manifestation) |
O → PD |
9.2 |
| 15 |
M/73 |
yes |
no |
n.d. |
no |
3 cycles tagraxofusp |
O*** → CR |
17.7* |
| 16 |
M/68 |
yes |
no |
no |
no |
AUTO-SCT (hyper-CVAD) |
AUTO → CR |
87.3 |
| 17 |
M/83 |
yes |
no |
no |
no |
None (only skin manifestation) |
O → PD |
56.5 |
| 18 |
F/67 |
yes |
no |
no |
no |
AUTO-SCT (hyper-CVAD) |
AUTO → CR |
110.6** |
| 19 |
M/56 |
yes |
yes |
yes |
yes |
ALLO (GRAALL-2014B) |
ALLO → CR |
79.6* |
| 20 |
M/53 |
no |
yes |
yes |
yes |
ALLO-SCT (Hyper-CVAD) |
ALLO → CR → Relapse |
4.5 |
| 21 |
M/70 |
yes |
yes |
yes |
yes |
5 cycles tagraxofusp |
O*** → PR → Progress |
11.9 |
| 22 |
M/21 |
yes |
yes |
yes |
yes |
1 cycle tagraxofusp |
O*** → PD → ALLO |
33.1* |
| 23 |
M/69 |
yes |
yes |
yes |
no |
Prednisone/cyclophosphamide, 2 cycles
Tagraxofusp |
O*** → CR → Relapse |
9.6 |
| 24 |
M/53 |
yes |
no |
yes |
no |
ALLO (3 cycles tagraxofusp) |
ALLO → CR → Relapse |
17.7* |
| 25 |
M/82 |
yes |
yes |
yes |
yes |
AML (venetoclax, azacytidine) |
AML → PD |
13.9 |
| 26 |
M/78 |
yes |
no |
yes |
yes |
5 cycles tagraxofusp |
O*** → CR → Relapse |
15.8 |
Two examples of patient cases are described in figure 1. The median age of patients
was 68.5
years (range: 20–83). Out of these patients, 23 (89%) were male and 3 female.
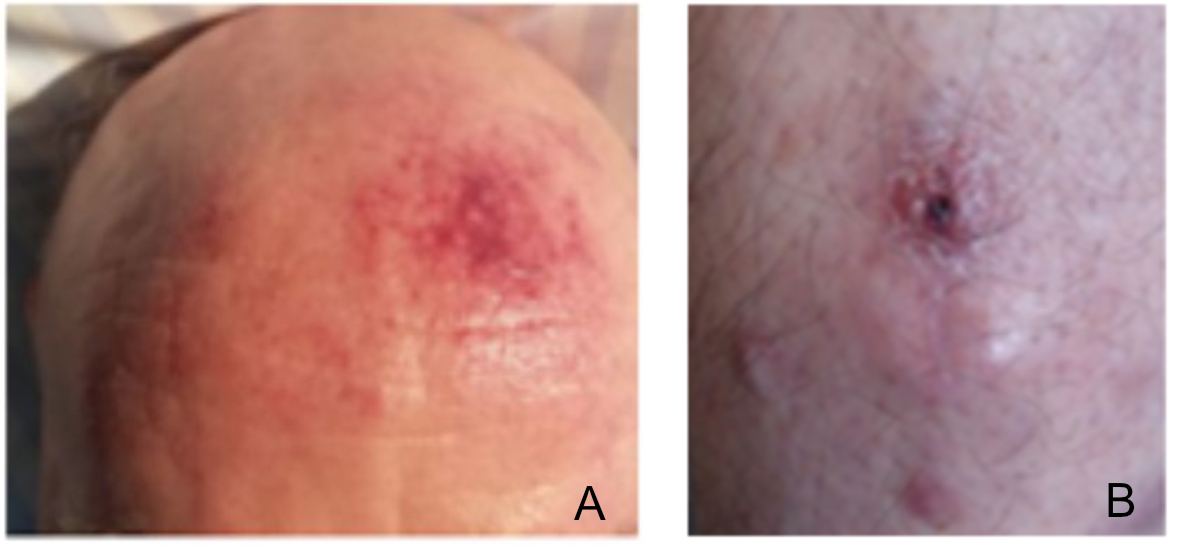
Figure 1Examples of two Swiss patients. (A) Patient #11: this
72-year-old man presented with affection of skin, lymph nodes, bone marrow,
peripheral blood and spleen. He underwent treatment with autologous stem cell
transplantation according to GRAALL-2012 protocol and achieved complete
remission. (B) Patient #14: This 71-year-old man initially presented
with a 6-month history of skin lesions. Based on the clinical and laboratory
findings, the patient was diagnosed with blastic plasmacytoid dendritic cell neoplasm
with
only skin manifestations. Due to age and comorbidities, allogeneic stem cell
transplantation was no option. Therefore, a “wait and see” approach was
adopted. Only one month later transformation into acute leukaemia was observed.
Chemotherapy with B-CHOP (bortezomibe, cyclophosphamide, vincristine,
adriablastine, prednisolone) was initiated, resulting in a very short partial
response and early disease progression, leading to introduction of a 2nd
line therapy with ABVD. Unfortunately, the patient passed away due to
complications from an intracerebral bleeding, 9 months after the diagnosis and
15 months after the onset of the first clinical symptoms.
Skin
manifestations at diagnosis were observed in 21 patients (81%), lymphadenopathy
in 12 patients (46%), bone marrow involvement in 17 patients (65%), and blasts
were detected in the peripheral blood in 11 cases (42%). Given the substantial
male predominance of our cohort, no conclusions regarding sex-based differences
could be drawn.
Ten
patients (38%) received a stem cell transplantation as first-line therapy (seven
ALLO and three AUTO). Of the seven patients who underwent ALLO, the induction
regimens were as follows: 3 AML-like, 1 ALL-like, 2 lymphoma-like and 1 Other (tagraxofusp).
The remaining
16 patients received 1st line treatment as follows: 1 ALL, 3 AML, 3 LL, 9 Other
(7 tagraxofusp, 2 Watch -and-Wait).
In brief,
the responses achieved by the seven patients with tagraxofusp plus one patient
with tagraxofusp as induction therapy before ALLO were as follows:
Patient #10
achieved a complete remission after only 3 cycles without further therapy. He
relapsed approximately 8 months after diagnosis and died due
to COVID-19 shortly after.
Patient #12
received an induction with 1 cycle of high-dose cytarabine, followed by 2
cycles of tagraxofusp. Despite achieving a complete remission after the 1st
cycle of tagraxofusp, he then progressed and died without further therapy.
Patient #15
received 3 cycles of tagraxofusp – complicated by severe capillary leak
syndrome – and achieved a complete remission. Without further treatment he was alive
with overall survival 17.7 months at last contact.
Patient #21
achieved a PR after 5 cycles of tagraxofusp. As treatment had to be stopped for
healthcare-insurance issues, he rapidly progressed and died without further
therapy.
Patient #22
progressed after 1 cycle of tagraxofusp, was stabilised with 2 cycles of
Hyper-CVAD and then underwent an ALLO. He was still alive 33 months after
diagnosis.
Patient #23
received 2 cycles of tagraxofusp after short induction therapy with
cyclophosphamide and prednisone, achieving a complete remission after one
cycle. Due to progression after the 2nd cycle, salvage therapy with azacytidine
and venetoclax was started. The patient died after the 4th cycle.
Patient #24
received 3 cycles of tagraxofusp as induction before ALLO which resulted in a complete
remission. Seven months later, he relapsed and was treated with Hyper-CVAD
before emigrating to his homeland. He was still alive 6 months after starting
salvage therapy.
Patient #26
achieved a complete remission after 3 cycles of tagraxofusp. After 5 cycles, he
progressed and was treated with seven cycles of azacytidine and venetoclax,
dying thereafter due to refractory disease.
Two other cases
deserve special attention, as they presented with asymptomatic skin lesions
only, leading to an initial Watch-and-Wait approach. Patient #17 was diagnosed
in 2011 at the age of 83 years. Four years later, he transformed to AML,
salvage therapy with hydroxyurea and later decitabine was initiated without
effect and the patient passed away. Patient #14 is described in figure 1.
All seven patients
who underwent ALLO as first-line along with the three patients who underwent AUTO
achieved complete remission, compared to only 38% (n = 6) of the 16 patients
without transplantation (p = 0.009). Considering overall survival, 1st line
therapy with AUTO/ALLO had much better outcomes than the rest (HR: 0.1, 95% CI:
0.02–0.47, p <0.001, figure 2) with a median overall survival of 86 months (4.5–NR)
vs 9.8 (6.1–13.8) months.
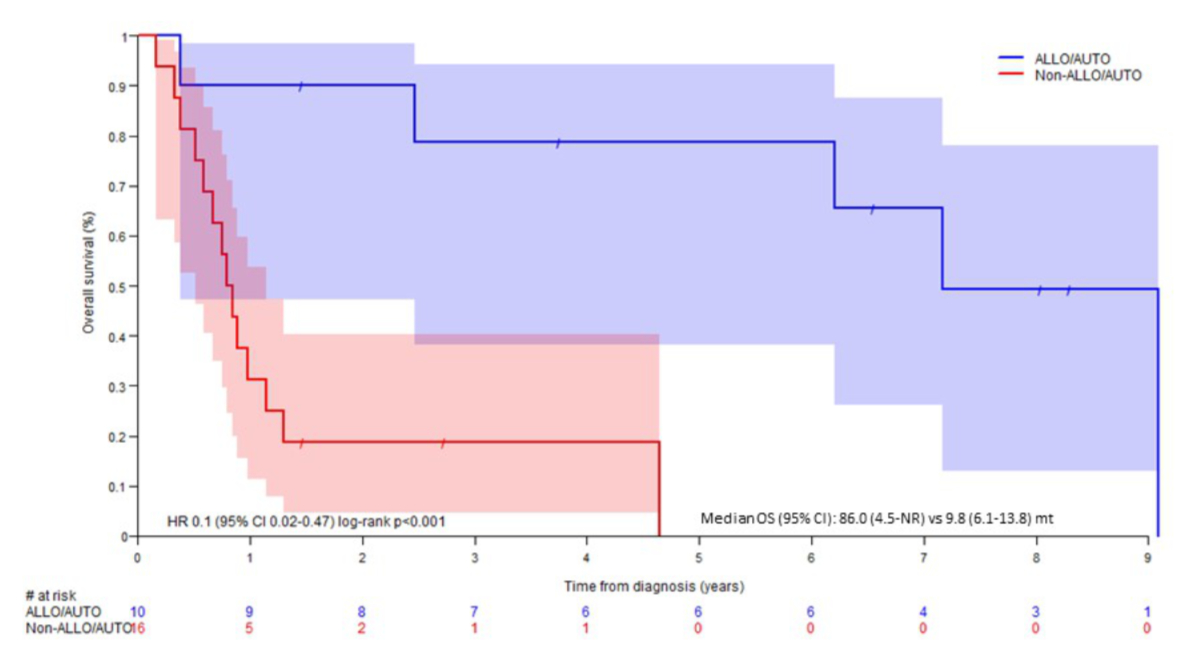
Figure 2Kaplan-Meier curve of overall survival in the Swiss cohort according
to therapy.
The seven
patients still alive were treated with ALLO (four patients, one of them with
tagraxofusp – induction), tagraxofusp (two patients, 1 of them progressed and underwent
ALLO as 2nd line) and AUTO (one patient).
The median
follow-up time was 8 years. The median overall survival was 1.2 years (95% CI:
0.8–6.2 years) (figure 3). The multivariate analysis revealed a significant
association between survival and involvement of skin (HR: 0.22, 95% CI: 0.06–0.82,
p = 0.024) and 1st line therapy (ALLO/AUTO vs non-ALLO/AUTO, HR: 0.06, 95% CI:
0.01–0.32, p = 0.001). Patients with skin involvement at diagnosis and
treatment with ALLO/AUTO achieved a particularly good outcome (small numbers,
data not shown).
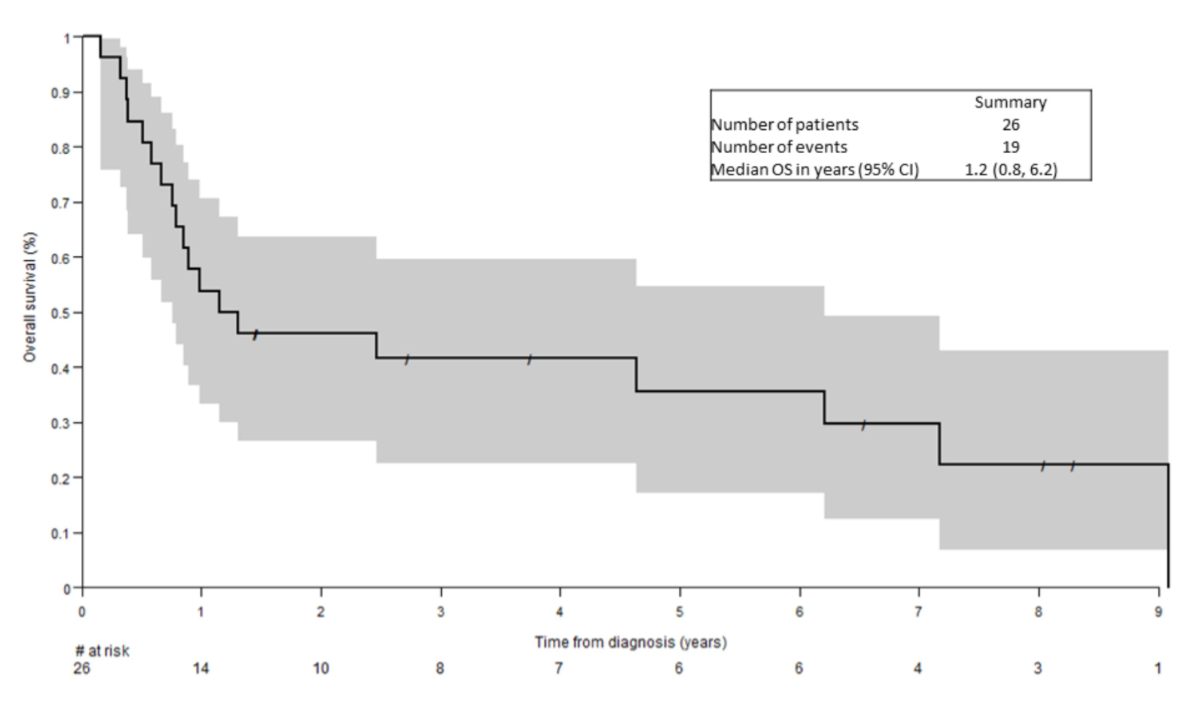
Figure 3Kaplan-Meier curve of overall survival in the Swiss cohort.
Structured review
of the literature
We
identified 115 international studies (listed in the appendix) including
600 patients from 1997 to 2020.
Most of the
studies were case reports with fewer than ten patients. Details regarding
therapeutic protocols and outcomes are lacking in many studies with larger
cohorts [11, 14–22].
Most
included patients were Europeans (n = 385, 64%) and Asians (n = 120, 20%),
followed by Americans (n = 90, 15%) and patients from Australia/New Zealand (n
= 3) and Africa (n = 2). Of the 115 publications, 96 (148 patients) were case
reports and 18 (405 patients) retrospective cohort analyses. We found only one
(47 patients) prospective study (the first phase 1/2 trial with tagraxofusp [13]).
BPDCN was
more common in men with a predominance of 3:1. The median age (n = 414) at
diagnosis was 66.5 years (ranging from 1 month to 103 years). Of note, women
were significantly younger than men (median: 62 vs 67 years, mean: 53.4 vs 59.3
years, p = 0.027).
The
clinical picture at diagnosis was heterogeneous (n = 516): skin lesions were
observed in 249 patients (missing data in 219 patients), lymphadenopathy in 94
(missing data in 309 patients), bone marrow infiltration in 116 (missing data
in 309 patients) and blasts in peripheral blood as an expression of leukaemic
state in 47 (missing data in 309 patients). Based on sex, there were significant
differences in the clinical presentation regarding bone marrow infiltration (p =
0.005) and lymph node involvement (p = 0.022) with both being more common in
males (table 3).
Table 3Differences in presentation by sex.
|
Females (n = 119) |
Males (n = 295) |
Missing
data |
Fisher’s exact test |
| Involved organ |
n (%) |
n (%) |
n |
p-value (2-tailed) |
| Bone marrow |
25/57 (43.9%) |
91/148 (61.5%) |
209 |
0.028 |
| Lymph nodes |
19/59 (32.2%) |
75/147 (51.0%) |
208 |
0.02 |
| Peripheral blood (blasts) |
9/58 (15.5%) |
38/147 (25.9%) |
209 |
0.141 |
| Skin |
71/87 (81.6%) |
178/210 (84.8%) |
117 |
0.493 |
Other
manifestations affecting <10% of patients included hepatomegaly (7.8%),
splenomegaly (9.1%), central nervous system involvement (5.4%), or lung and
pleural involvement (1.9%). However, almost any organ could be involved in
individual cases (such as chest wall, gastrointestinal tract, conjunctiva,
testis, buttock, ovaries, paravertebral tissue, parotid gland, lacrimal gland
and thyroid gland, gallbladder, orbital cavity).
Unfortunately,
we found no / very little data on the time elapsed between the onset of the
first symptoms and the diagnosis.
We tried to
assign each of the
516 patients to a therapeutic regimen (figure 4). ALLO was more prevalent in
Europe (18% of European patients), Asia and America. Most patients in America
were treated with ALL, while AML was predominantly performed in Europe. LL were
in use in all continents in about 15–25% of patients. As (unlike in the Swiss
Cohort) the primary attribution could not be undertaken with sufficient
certainty, only 357 patients were considered for demonstrating the distribution
of different 1st line therapeutic regimens. The most used regimens were ALLO (87
or 24%), followed by LL (83 or 23%), ALL (49 or 14%), AML (28 or 8%), AUTO (15 or
4%) and AL NOS (11 or 3%). A quarter of patients (84 or 24%) received a
different treatment (“Other” category). Survival data were available for 364
patients. The median overall survival was 15 months (95% CI: 13–18, figure 5A)
with no significant differences between sexes (p = 0.13, figure 5B).
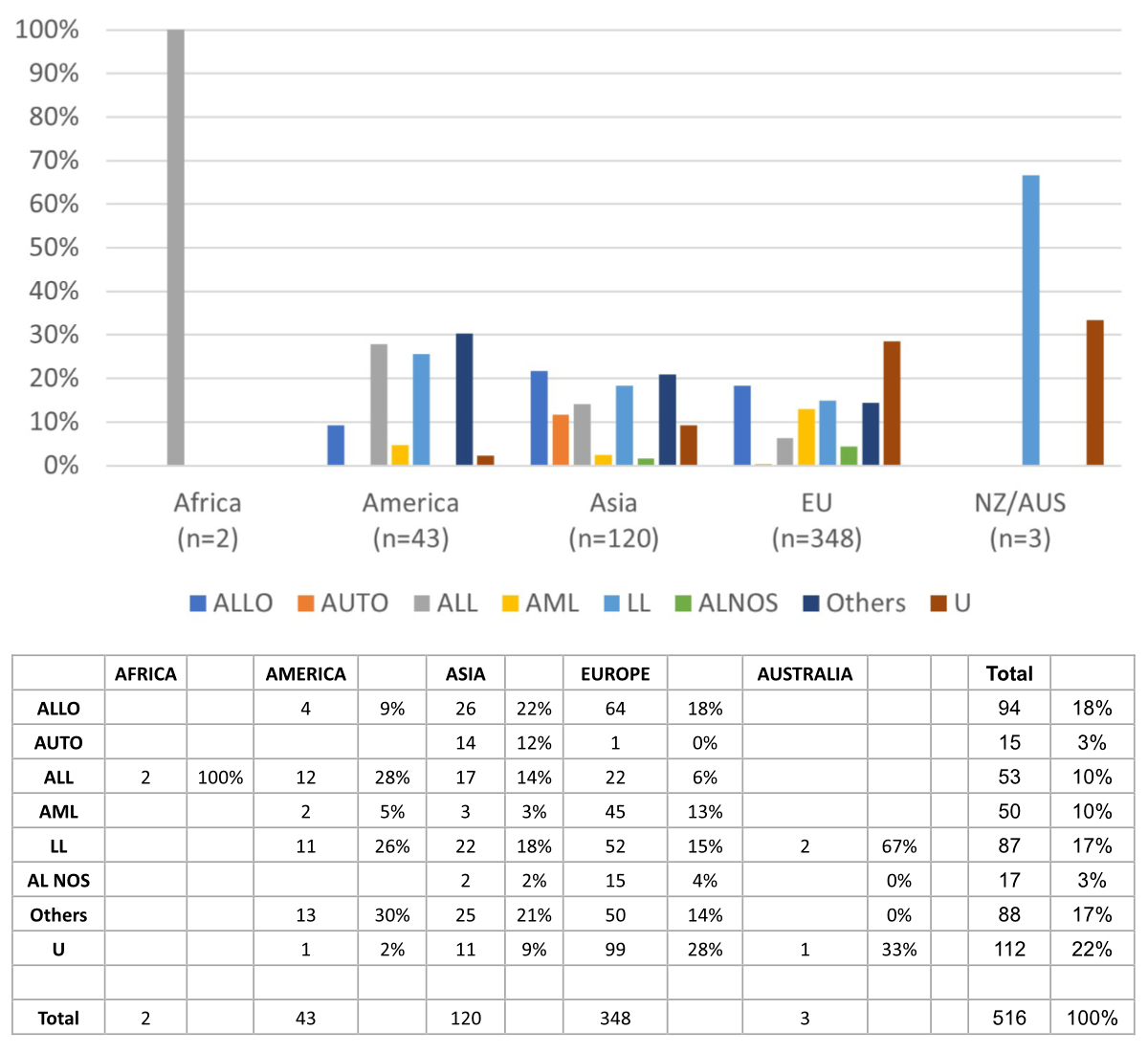
Figure 4Geographic distribution of therapeutic regimens. U: unknown.
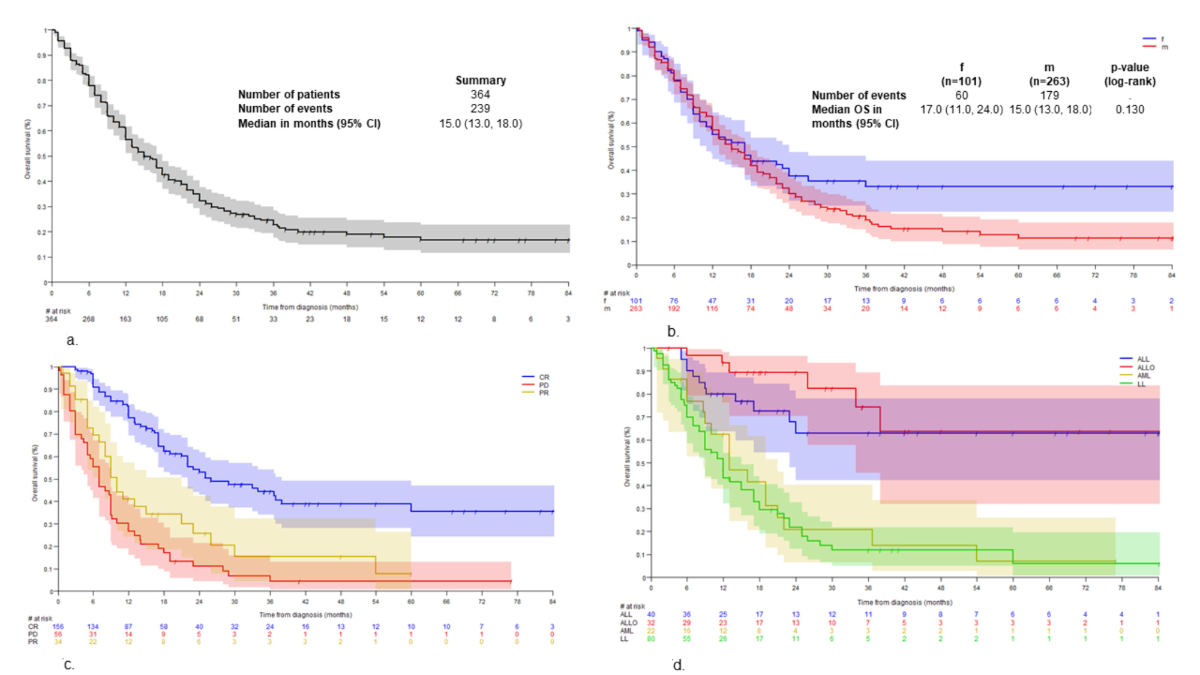
Figure 5Overall survival according to different conditions. (A)
Overall survival – structured review of the literature; (B) Overall
survival according to sex; (C) Overall survival according to remission
status; (D) Overall survival according to therapeutic regimens.
Achieving a
complete remission was significantly associated with a longer overall survival
(figure 5C). Upfront transplant-concepts resulted in a high complete remission rate,
e.g. ALLO vs non-ALLO (71.3% vs 57.8%, p <0.001) and ALLO/AUTO vs
non-ALLO/AUTO (78.6% vs 26.0%, p = 0.001), ALL vs non-ALL (81.6% vs 57.8%, p = 0.026).
AUTO vs non-AUTO was not meaningful due to small sample size. Overall, more
intensive approaches like ALLO or ALL were associated with a longer overall
survival (figure 5D; only therapeutic regimens with sufficient sample sizes are
considered). ALL was non-inferior to ALLO in terms of overall survival (p = 0.416).
In a univariate
analysis, overall survival was determined by age, involvement of bone marrow
and other organs, blasts in the peripheral blood, choice of 1st line therapy
and response to 1st line therapy (table 4).
Table 4Univariate analysis of parameters influencing overall survival.
| Variable |
n |
HR (95% CI) |
p-value |
| Age (years) |
414 |
1.03 (1.02–1.04) |
<0.001 |
| Sex (M vs F) |
414 |
1.25 (0.93–1.68) |
0.134 |
| Skin (Yes vs No) |
297 |
1.02 (0.67–1.56) |
0.922 |
| Lymph
nodes (Yes vs No) |
206 |
1.45 (0.97–2.15) |
0.068 |
| Bone
marrow (Yes vs No) |
205 |
1.53 (1.02–2.31) |
0.042 |
| Peripheral
blood (Yes vs No) |
205 |
1.72 (1.11–2.67) |
0.016 |
| Other
organ involvement (Yes vs No) |
206 |
1.66 (1.07–2.56) |
0.023 |
| Complete
remission (Yes vs No) |
349 |
0.33 (0.25–0.44) |
<0.001 |
| Relapse (Yes vs No) |
193 |
1.45 (0.98–2.14) |
0.060 |
| Leukaemic
transformation (Yes vs No) |
155 |
1.74 (0.99–3.05) |
0.054 |
| Variable (1st line treatment) |
| ALLO/AUTO vs non-ALLO/AUTO |
254 |
0.18
(0.09–0.39) |
<0.001 |
| ALLO/AUTO/ALL vs non-ALLO/AUTO/ALL |
254 |
0.19
(0.11–0.30) |
<0.001 |
| ALLO vs non-ALLO |
254 |
0.21
(0.10–0.45) |
<0.001 |
| ALL/AUTO
vs non-ALL/AUTO |
254 |
0.27
(0.15–0.49) |
<0.001 |
| ALL vs non-ALL |
254 |
0.31
(0.17–0.55) |
<0.001 |
The multivariate
analysis showed that in addition to the treatment concept, achieving a complete
remission in first line was the strongest additional prognostic factor, and
that bone marrow infiltration at diagnosis had a negative impact on overall
survival (table 5).
Table 5Multivariate analysis of parameters influencing overall
survival.
| Variable |
HR (95% CI) |
p-value |
| Complete
remission (yes vs no) |
0.06 (0.02–0.18) |
<0.001 |
| ALLO/AUTO vs
non-ALLO/AUTO |
0.09 (0.02–0.38) |
<0.001 |
| Relapse (yes vs no) |
8.63 (2.89–25.76) |
<0.001 |
| Bone
marrow (yes vs no) |
2.24 (1.28–3.92) |
0.005 |
| Age (years) |
1.01 (1.00–1.02) |
0.094 |
| Lymph
nodes (yes vs no) |
1.44 (0.86–2.40) |
0.163 |
| Peripheral
blood (yes vs no) |
1.20 (0.60–2.39) |
0.606 |
| Leukaemic transformation (yes vs no) |
1.19 (0.56–2.53) |
0.658 |
Discussion
Blastic
plasmacytoid dendritic cell neoplasm (BPDCN) is a very rare disease with an often
dismal outcome due to prolonged time to diagnosis, and lack of effective therapies
and of randomised controlled prospective trials. We expect around 3-4 new
diagnoses in Switzerland every year. The available literature mainly consists
of case reports, case series and retrospective analyses. Phase 3 trials are
lacking because of the rarity of the disease and the absence of recognised
therapeutic options. The clinical presentation is heterogeneous, with cutaneous
lesions often serving as a key to diagnosis, followed by lymphadenopathies,
bone marrow involvement, cytopenia and leukaemic transformation.
Our cohort
of Swiss patients contained 26 patients (two additional patients were not included
in the analyses due to lack of informed consent) in 18 years with a median follow-up
of 8 years. The mean population in
Switzerland during this period was 8.1 million, yielding an incidence of 0.19 /
million / year, which is slightly less than expected. The median age of onset,
the male predominance, clinical presentations and survival curves are in line
with previous findings as demonstrated in our structured literature review (see
below).
The rate of
patients undergoing allogeneic transplantation is similar (Switzerland: 31%, structured
review of the literature: 24%), as is the general heterogeneity of therapeutic
regimens in use. Due to the small sample size, our findings were not
statistically significant. However, we made some interesting observations. All seven
patients with ALLO as first line achieved a complete remission; the same holds
true for the 3 AUTO patients. Only one ALL-regimen upfront was chosen in
Switzerland (PD). Of 3 LL, only one led to a complete remission (1 PR, 1 PD)
and all three AML (without consolidating ALLO) resulted in PD. The nine other first-line
strategies mainly contained tagraxofusp (seven patients; 5 complete remission, 1
PR, 1 PD) or Watch-and-Wait (because of modest skin manifestation only, n = 2).
Nearly all patients (with two exceptions) diagnosed in 2020 or later had tagraxofusp
as part of their treatment. The follow-up is still too short to comment on the
efficacy of tagraxofusp. The treatment regimen in the relapsed setting
contained hydroxyurea, ALL protocols, B-CHOP, ABVD, hypomethylating agents and
venetoclax. Of the 26 patients, seven are still alive (4 ALLO, 1 AUTO, 1
tagraxofusp) including one patient with ALLO 2nd line after progression to
tagraxofusp and salvage treatment with Hyper-CVAD.
The structured
review of the literature confirmed that BPDCN is mainly a disease of older age,
even if it can occur at any age. Male patients seemed to more often have bone
marrow infiltration and lymphadenopathies at diagnosis than women, a fact not
affecting the outcome in a statistically significant manner. Median overall
survival was around 15 months. Assuming underdiagnosing and underreporting of
BPDCN, overall survival might even be shorter. Subdividing the therapeutic
approaches into six different groups, upfront allogenic concepts or ALL regimens
clearly performed better than AML-like or lymphoma-like treatments. The same
probably holds true for an upfront autologous concept, even if the numbers were
too small to draw firm conclusions. For the latter reason, the group with Other
treatments could not be subdivided and no serious evaluation of efficacy was
possible. Our study confirms that deeper responses to first-line therapy are
cornerstones of a better outcome with patients in complete remission achieving
the longest overall survival.
Different
new therapeutic strategies involving targeted therapies have been increasingly used
over the last decade. Bortezomib, an inhibitor of the nuclear factor-kappa B
pathway, demonstrated promising results in combination with chemotherapy in
mice [23]. The combination of lenalidomide and dexamethasone was successfully used
in several case reports [24]. In a case at our institution, the use of
bortezomib in combination with CHOP did not result in long-term remission.
The
anti-CD38 antibody daratumumab was also used with mixed results, since CD38 is
not only expressed on lymphoid tissues (mainly plasma cells), but also on
myeloid cells [25]. Immunohistochemical investigations of BPDCN biopsies revealed
[25] prominent staining of the anti-apoptotic protein BCL-2 leading to trials
with venetoclax, with no measurable success. The discovery of PD-L1 positivity in
approximately 50% of investigated BPDCN samples (range: 1% to 55%) has raised
hopes about the potential of checkpoint inhibitors against PD-L1, although no
such reports have been published to date [27]. Single reports of anti-CD123-CAR
T-cell therapy have been published with discordant outcomes [28].
Tagraxofusp
entered the scene a few years ago as the “new kid on the block”. This CD123-directed
cytotoxin led to a high ORR, serving as a perfect induction – or bridging – therapy
to transplant in eligible patients. A relevant and potentially lethal side
effect is capillary leak syndrome. Nevertheless, tagraxofusp has become a new
standard of care for patients with BPDCN and was approved by the Swiss
authorities (Swissmedic) by January 2024. As of today, allogenic stem cell
transplantation seems to be the best therapeutic option in patients with BPDCN,
especially in younger patients with a good performance status. Complete
remission can be achieved in about 60% of these patients. Future directions
will probably focus on the development of more appropriate (preferably
allogenic) CAR T-cell therapy or bispecific antibodies, as well as on
combination therapies including tagraxofusp.
The
strength of our study is the extensive work-up of available literature, and
this not only in the English language, but also in German, French and Italian.
It gives a comprehensive overview of BPDCN as a rare disease entity in the
national and worldwide context. By including the largest haemato-oncology centres
in Switzerland, a substantiated statement about prevalence and therapeutic
modalities in Switzerland can be given.
Our study
has several limitations. First, the data drawn from the 115 international
papers were often not complete, not related to the scope of the present study
and of heterogeneous quality. Since, as mentioned, the term BPDCN was only
introduced in 2008, it cannot be ruled out that we would have found further
(especially older) relevant literature with extended search terms. As such, stringent
conclusions beyond a descriptional and hypothesis-generating manner cannot be
drawn from this kind of even structured literature review. Secondly, due to
upcoming availability of tagraxofusp, original articles and case reports as
well as cohort studies were only considered up to 2020. Thirdly, collecting the
data for our Swiss cohort in a retrospective manner meant that not every centre
could provide us with required details, for example the time from first
symptoms to diagnosis and treatment. Further, it cannot be excluded that a few
BPDCN patients were treated outside the larger centres, even if the probability
is low. As the number of patients in the Swiss cohort is very small, it is
difficult to demonstrate statistically significant differences between
subgroups.
In
conclusion, our study yielded deeper insights into a rare, heterogeneous
disease, which is difficult to treat and where comparative studies are lacking.
We propose that national or even continental registries be established for
every orphan disease.
Acknowledgments
The authors
would like to thank the patients and their families for agreeing to register
within the Swiss cohort.
Adrian
Schmidt, MD
Department of Internal Medicine
Clinic for Medical
Oncology and Hematology
Municipal Hospital Zurich Triemli
Birmensdorferstrasse 497
CH-8063 Zurich
adrian.schmidt[at]stadtspital.ch
References
1. Guru Murthy GS, Pemmaraju N, Atallah E. Epidemiology and survival of blastic plasmacytoid
dendritic cell neoplasm. Leuk Res. 2018 Oct;73:21–3. doi: https://doi.org/10.1016/j.leukres.2018.08.014
2. Facchetti F. Ungari M, Marocolo D, Lonardi S, Vermi W. Blastic plasmacytoid dendritic
cell neoplasm. Hematology Meeting Reports. 2009;3(3):1–3. 10.4081/hmr.v3i3.553
3. Herling M, Jones D. CD4+/CD56+ hematodermic tumor: the features of an evolving entity
and its relationship to dendritic cells. Am J Clin Pathol. 2007 May;127(5):687–700.
doi: https://doi.org/10.1309/FY6PK436NBK0RYD4
4. Chaperot L, Bendriss N, Manches O, Gressin R, Maynadie M, Trimoreau F, et al. Identification
of a leukemic counterpart of the plasmacytoid dendritic cells. Blood. 2001 May;97(10):3210–7.
doi: https://doi.org/10.1182/blood.V97.10.3210
5. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision
of the World Health Organization classification of lymphoid neoplasms. Blood. 2016 May;127(20):2375–90.
doi: https://doi.org/10.1182/blood-2016-01-643569
6. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition
of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid
and Histiocytic/Dendritic Neoplasms. Leukemia. 2022 Jul;36(7):1703–19. doi: https://doi.org/10.1038/s41375-022-01613-1
7. Pemmaraju N, Kantarjian H, Sweet K, Wang E, Senapati J, Wilson NR, et al.; North American
Blastic Plasmacytoid Dendritic Cell Neoplasm Consortium. North American Blastic Plasmacytoid
Dendritic Cell Neoplasm Consortium: position on standards of care and areas of need.
Blood. 2023 Feb;141(6):567–78. doi: https://doi.org/10.1182/blood.2022017865
8. Marco Herling PB. Antonio Cozzio, Edgar Dippel, Peter Dreger, Emmanuella Guenova,
Constanze Jonak, Markus G. Manz, Ilske Oschlies, Peter Reimer, Andreas Rosenwald,
Ingrid Simonitsch-Klupp, Bernhard Wörmann. [updated January 2022]; Available from:
www.onkopedia.com/de/onkopedia/guidelines/blastische-plasmazytoide-dendritische-zellneoplasie-bpdcn/@@guideline/html/index.html
9. Pollyea DA, Altman JK, Assi R, Bixby D, Fathi AT, Foran JM, et al. Acute Myeloid Leukemia,
Version 3.2023, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw.
2023 May;21(5):503–13. 10.6004/jnccn.2023.0025
10. Julia F, Dalle S, Duru G, Balme B, Vergier B, Ortonne N, et al. Blastic plasmacytoid
dendritic cell neoplasms: clinico-immunohistochemical correlations in a series of
91 patients. Am J Surg Pathol. 2014 May;38(5):673–80. doi: https://doi.org/10.1097/PAS.0000000000000156
11. Pagano L, Valentini CG, Pulsoni A, Fisogni S, Carluccio P, Mannelli F, et al.; GIMEMA-ALWP
(Gruppo Italiano Malattie EMatologiche dell’Adulto, Acute Leukemia Working Party).
Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: an Italian
multicenter study. Haematologica. 2013 Feb;98(2):239–46. doi: https://doi.org/10.3324/haematol.2012.072645
12. Lee JK, Schiller G. Blastic plasmacytoid dendritic cell neoplasm. Clin Adv Hematol
Oncol. 2012 Jan;10(1):60–2.
13. Pemmaraju N, Lane AA, Sweet KL, Stein AS, Vasu S, Blum W, et al. Tagraxofusp in Blastic
Plasmacytoid Dendritic-Cell Neoplasm. N Engl J Med. 2019 Apr;380(17):1628–37. doi: https://doi.org/10.1056/NEJMoa1815105
14. Julia F, Petrella T, Beylot-Barry M, Bagot M, Lipsker D, Machet L, et al. Blastic
plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol.
2013 Sep;169(3):579–86. doi: https://doi.org/10.1111/bjd.12412
15. Feuillard J, Jacob MC, Valensi F, Maynadié M, Gressin R, Chaperot L, et al. Clinical
and biologic features of CD4(+)CD56(+) malignancies. Blood. 2002 Mar;99(5):1556–63.
doi: https://doi.org/10.1182/blood.V99.5.1556
16. Roos-Weil D, Dietrich S, Boumendil A, Polge E, Bron D, Carreras E, et al.; European
Group for Blood and Marrow Transplantation Lymphoma, Pediatric Diseases, and Acute
Leukemia Working Parties. Stem cell transplantation can provide durable disease control
in blastic plasmacytoid dendritic cell neoplasm: a retrospective study from the European
Group for Blood and Marrow Transplantation. Blood. 2013 Jan;121(3):440–6. doi: https://doi.org/10.1182/blood-2012-08-448613
17. Tsagarakis NJ, Kentrou NA, Papadimitriou KA, Pagoni M, Kokkini G, Papadaki H, et al.;
Hellenic Dendritic Cell Leukemia Study Group. Acute lymphoplasmacytoid dendritic cell
(DC2) leukemia: results from the Hellenic Dendritic Cell Leukemia Study Group. Leuk
Res. 2010 Apr;34(4):438–46. doi: https://doi.org/10.1016/j.leukres.2009.09.006
18. Hashikawa K, Niino D, Yasumoto S, Nakama T, Kiyasu J, Sato K, et al. Clinicopathological
features and prognostic significance of CXCL12 in blastic plasmacytoid dendritic cell
neoplasm. J Am Acad Dermatol. 2012 Feb;66(2):278–91. doi: https://doi.org/10.1016/j.jaad.2010.12.043
19. Dalle S, Beylot-Barry M, Bagot M, Lipsker D, Machet L, Joly P, et al. Blastic plasmacytoid
dendritic cell neoplasm: is transplantation the treatment of choice? Br J Dermatol.
2010 Jan;162(1):74–9. doi: https://doi.org/10.1111/j.1365-2133.2009.09373.x
20. Aoki T, Suzuki R, Kuwatsuka Y, Kako S, Fujimoto K, Taguchi J, et al. Long-term survival
following autologous and allogeneic stem cell transplantation for blastic plasmacytoid
dendritic cell neoplasm. Blood. 2015 Jun;125(23):3559–62. doi: https://doi.org/10.1182/blood-2015-01-621268
21. Brüggen MC, Valencak J, Stranzenbach R, Li N, Stadler R, Jonak C, et al. Clinical
diversity and treatment approaches to blastic plasmacytoid dendritic cell neoplasm:
a retrospective multicentre study. J Eur Acad Dermatol Venereol. 2020 Jul;34(7):1489–95.
doi: https://doi.org/10.1111/jdv.16215
22. Cernan M, Szotkowski T, Hisemova M, Cetkovsky P, Sramkova L, Stary J, et al. Blastic
plasmacytoid dendritic cell neoplasm: first retrospective study in the Czech Republic.
Neoplasma. 2020 May;67(3):650–9. doi: https://doi.org/10.4149/neo_2020_190507N407
23. Philippe L, Ceroi A, Bôle-Richard E, Jenvrin A, Biichle S, Perrin S, et al. Bortezomib
as a new therapeutic approach for blastic plasmacytoid dendritic cell neoplasm. Haematologica.
2017 Nov;102(11):1861–8. doi: https://doi.org/10.3324/haematol.2017.169326
24. Marmouset V, Joris M, Merlusca L, Beaumont M, Charbonnier A, Marolleau JP, et al. The
lenalidomide/bortezomib/dexamethasone regimen for the treatment of blastic plasmacytoid
dendritic cell neoplasm. Hematol Oncol. 2019 Oct;37(4):487–9. doi: https://doi.org/10.1002/hon.2671
25. Iversen KF, Holdgaard PC, Preiss B, Nyvold CG, Plesner T. Daratumumab for treatment
of blastic plasmacytoid dendritic cell neoplasm. A single-case report. Haematologica.
2019 Sep;104(9):e432–3. doi: https://doi.org/10.3324/haematol.2018.214635
26. Montero J, Stephansky J, Cai T, Griffin GK, Cabal-Hierro L, Togami K, et al. Blastic
Plasmacytoid Dendritic Cell Neoplasm Is Dependent on BCL2 and Sensitive to Venetoclax.
Cancer Discov. 2017 Feb;7(2):156–64. doi: https://doi.org/10.1158/2159-8290.CD-16-0999
27. Aung PP, Sukswai N, Nejati R, Loghavi S, Chen W, Torres-Cabala CA, et al. PD1/PD-L1
Expression in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers (Basel). 2019 May;11(5):695.
doi: https://doi.org/10.3390/cancers11050695
28. Jiang YL, Li Q, Yuan T, Jiang YY, Deng Q. Case Report of Anti-CD123 Chimeric Antigen
Receptor T-Cell Therapy Followed by Radiotherapy for a Recurrence of Blastic Plasmacytoid
Dendritic Cell Neoplasm After Allogeneic Hematopoietic Stem Cell Transplantation.
OncoTargets Ther. 2020 Apr;13:3425–30. doi: https://doi.org/10.2147/OTT.S250016
Appendix
The appendix is available in the pdf version of the article at https://doi.org/10.57187/s.3885.